BRAIN VOLUMES
As with other areas of anti-cannabis legislation the
literature on attention deficit hyperactivity disorder is strewn with the debris
of officialdom not knowing what it did not know.
Pim Ittiphakorn, Simon Erridge, and Mikael Sodergren
of Imperial College London, James Rucker of Kings College London, and Carl
Holvey and Ross Coomber of Sapphire Medical Clinics, explain what we now believe
about cannabis and ADHD - "now" meaning 6 December 2023:
"Attention-deficit/hyperactivity disorder (ADHD) is
one of the most common psychiatric disorders, with an estimated global
prevalence of 5% in children and 2.5% in adults. The estimated incidence of ADHD
diagnosis has increased by approximately 42% in children between 2003 and 2011,
and 123% in adults between 2007 and 2016 in the United States. ADHD is
characterized by symptoms of inattentiveness, hyperactivity, and impulsiveness
causing functional impairment in two or more settings (e.g., work and home).
ADHD is often associated with psychosocial difficulties, such as relationship
problems, unemployment, educational underachievement, and criminality. Moreover,
ADHD is also associated with a higher incidence of sleep disturbance and
psychiatric co-morbidities, including anxiety, substance misuse, and depression.
As a result, these issues can significantly reduce the quality of life for
individuals with ADHD.
"Current treatment for ADHD consists of a combination
of psychological therapies and both stimulant and non-stimulant medications.
Stimulants are the most commonly prescribed medications for ADHD and target
executive and attentional function. They are considered relatively safe and
effective treatments, however, they are commonly associated with decreased
appetite, insomnia, emotional dysregulation, irritability, and an increased risk
of adverse cardiovascular events. Non-stimulant medications have been shown to
reduce ADHD-related functional impairments and co-occurring mood disorders.
Despite their effectiveness, medication adherence rates are relatively low due
to the adverse events that are commonly experienced. This highlights the need
for novel therapeutics for ADHD.
"The endocannabinoid system (ECS) plays a vital role
in cognitive function, motor coordination, and emotional homeostasis, in
addition to the regulation of dopaminergic pathways in the brain. The ECS is a
signaling network consisting of endocannabinoids, enzymes, and cannabinoid
receptors, including cannabinoid type 1 (CB1) receptors and cannabinoid type 2
(CB2) receptors. Dysregulation in the ECS has been implicated in the
pathophysiology of ADHD. CB1 receptors are widely distributed throughout the
central nervous system, with high levels found in regions associated with
cognitive functioning and processing, such as the basal ganglia, cerebellum,
neocortex, and hippocampus."
https://onlinelibrary.wiley.com/doi/10.1002/npr2.1240 [4264]
Although ADHD's roots as a concept can be traced back
to 1798, people didn't really believe in physiological bases for behavioural
disorders until a lot later.
In 2010 Klaus W Lange worked at the Department of
Biological and Abnormal Psychology, University of Regensburg. According to Lange
et al's "The history of attention deficit hyperactivity disorder" printed,
appropriately enough, in Attention Deficit Hyperactivity Disorder, it was in
1902 that "defective moral control" gained a semblance of scientific definition,
focussing on
"(1) passionateness; (2) spitefulness – cruelty; (3)
jealousy; (4) lawlessness; (5) dishonesty; (6) wanton mischievousness –
destructiveness; (7) shamelessness – immodesty; (8) sexual immorality; and (9)
viciousness. The keynote of these qualities is self-gratification, the immediate
gratification of self without regard either to the good of others or to the
larger and more remote good of self."
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3000907/ [1888]
George F Still attempts to wrestle with the evidence
using the tools available at the time, which basically amount to listening to
stories, a physical examination, and measuring the circumference of the troubled
kids' above-average sized heads:
"Another boy was brought to me at the age of five
years with a history that for two months he had been very excitable and had at
the same time become extremely spiteful, throwing things at people apparently in
wanton spitefulness and attacking strange children in the street without any
provocation ; he had expressed a wish one day to 'chop his mother’s head off
with a chopper,' and was caught one day in the act of putting the cat into the
fire, and on a subsequent occasion- he attempted to put it into a copper of
boiling water. I saw this boy 18 months later when he was said still to be very
excitable and extremely passionate, kicking or striking anyone who offended him.
Nine months previously he had hit his mother on the head with a big toy gun
because he could not have some trifling thing that he wanted; he was also said
to be spiteful to other children. He was untruthful, but his lying was of the
purely romantic type, so much so that it was difficult to imagine that the boy
intended to deceive. His head was unusually large, measuring 21⅜ inches in
maximum circumference at the age of six and a half years. He is a heavylooking
but well-grown boy and he is fully up to the average in school attainments. His
maternal grandfather had diabetes, one maternal uncle attempted suicide twice,
and two other maternal uncles have become confirmed drunkards. The boy’s parents
are respectable middle-class people and seemed to give the child excellent
care."
Dr Still noticed some things we would today ascribe to
other syndromes: the pronounced epicanthic folds perhaps of fetal alcohol
syndrome, or the repetitive actions of obsessive compulsive disorder. He looks
for suspected causes in the medical histories of family members. By today's
statistical standards, Still's collection of case histories is nothing more than
anecdotal. But it's from here the idea of a hyperkinetic disorder begins to take
shape, culminating in the new name attention deficit disorder (ADD) in 1980.
https://ia800708.us.archive.org/view_archive.php?archive=/22/items/crossref-pre-1909-scholarly-works/10.1016%252Fs0140-6736%252801%252970006-2.zip&file=10.1016%252Fs0140-6736%252801%252970022-0.pdf
[1889]
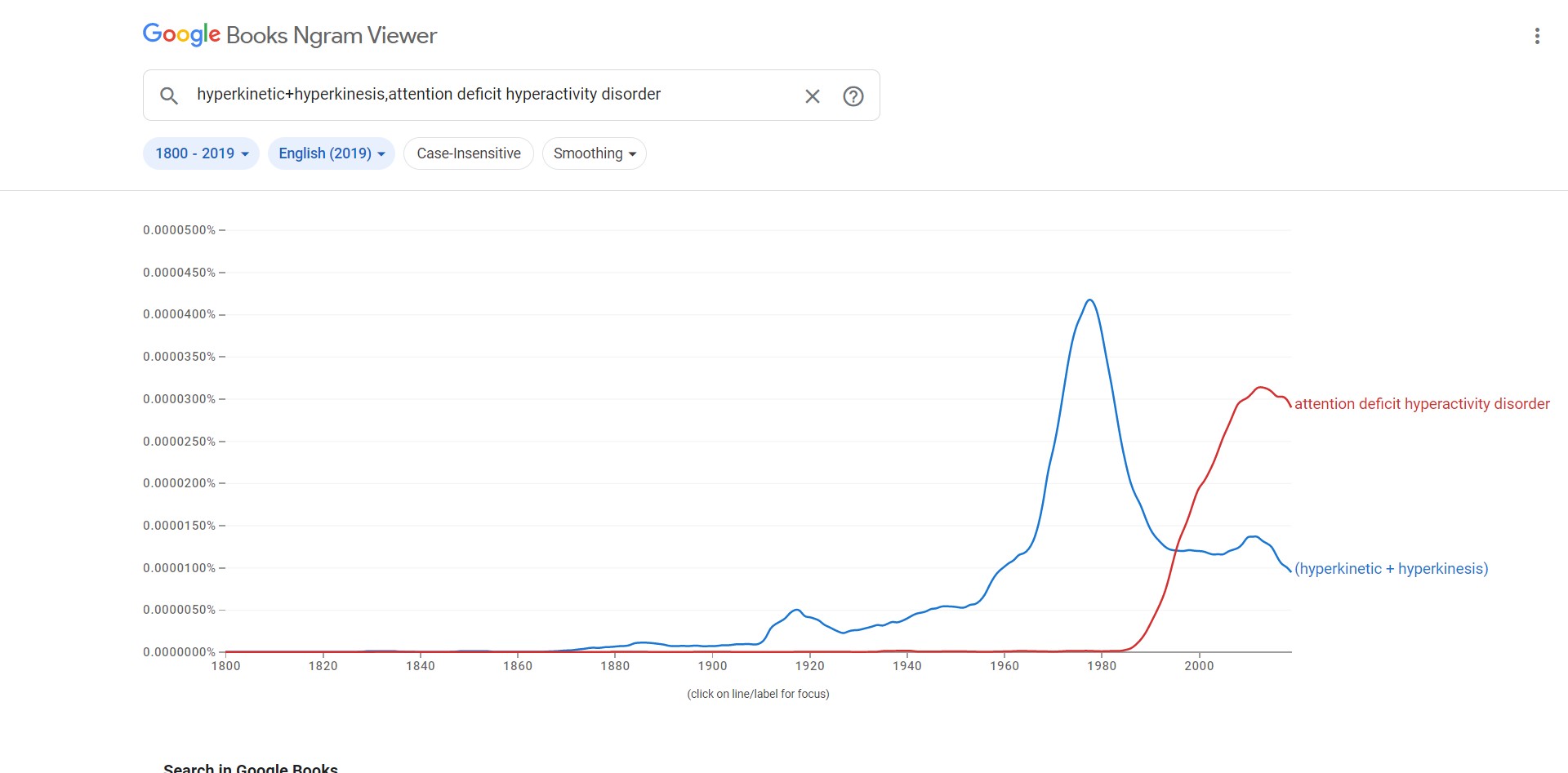
Here's how the nomenclature looks in the English
corpus
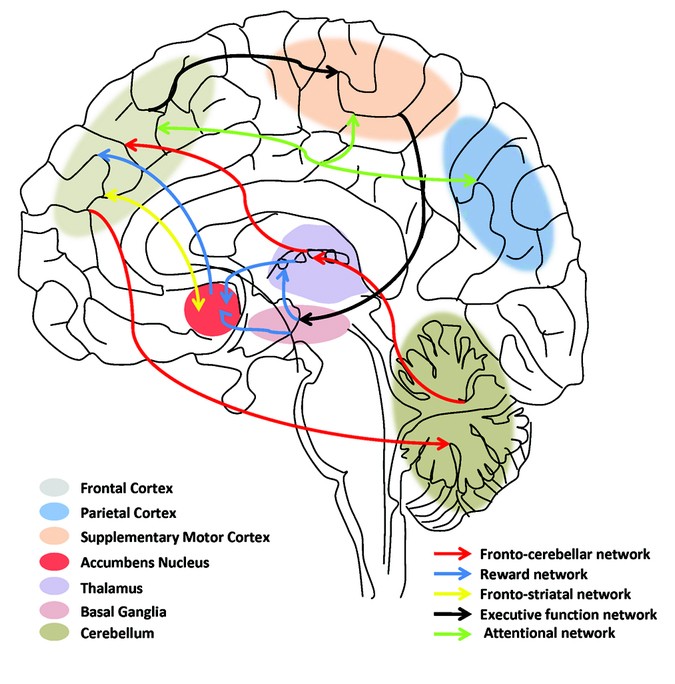
Here's a diagram to help explain the neurobiology of
ADHD, as it was understood in 2011.
According to these French authors,
"A study of pregnant mothers related or unrelated to
their child as a result of in vitro fertilization showed that prenatal stress
was linked to ADHD only when mothers were related to their child, suggesting
that the association may be accounted for by inherited factors. A recent twin
study focused on ADHD-related conditions (antisocial behavior and substance use
disorders in young adults), has provided an important insight into mechanisms of
gene-environment influence on externalizing disorders by showing that genetic
factors contribute more to the development of behavioral symptoms in a context
of high environmental adversity, in accordance with a diathesis-stress model.
These examples illustrate the importance of genetically informed study designs
to further disentangle environmental and genetic contributions to ADHD."
https://www.nature.com/articles/pr9201196 [2008]
From New York to Australia, Isik et al (2023) have
been looking at "Neurodevelopmental outcomes in children after prenatal
marijuana exposure"
"This study evaluated the association between PME and
neuropsychological test scores in late childhood and early adulthood, accounting
for a wide range of parental characteristics.
"Methods: This study evaluated participants from the
Raine Study, a cohort of 2868 children born between 1989 and 1992 [formerly
known as the West Australian Pregnancy Cohort Study]. Children whose mothers
provided information on marijuana use during pregnancy were included. The
primary outcome was the Clinical Evaluation of Language Fundamentals (CELF) at
age 10. Secondary outcomes included the Peabody Picture Vocabulary Test (PPVT),
Child Behaviour Checklist (CBCL), McCarron Assessment of Neuromuscular
Development (MAND), Coloured Progressive Matrices (CPM), Symbol Digit Modality
Test (SDMT) and Autism Spectrum Quotient (AQ) scores. Exposed and unexposed
children were matched by propensity score using optimal full matching. Missing
covariate data were imputed using multiple imputation. Inverse probability of
censoring weighting (IPCW) was used to adjust for missing outcome data. Linear
regression within matched sets, adjusted by IPCW, evaluated score differences
between exposed and unexposed children. As a secondary analysis, modified
Poisson regression, adjusted by match weights and IPCW, evaluated the risk of
clinical deficit in each outcome following PME.
"Results: Of the 2804 children in this cohort, 285
(10.2%) had PME. After optimal full matching and IPCW, exposed children scored
similarly on CELF Total (-0.33 points, 95% confidence interval [CI] -4.71,
4.05), Receptive (+0.65 points, 95% CI -4.08, 5.38) or Expressive (-0.53 points,
95% CI -5.07, 4.02). PME was not associated with secondary outcomes or risks of
clinical deficit in any neuropsychological assessments.
"Conclusions: After adjusting for sociodemographic and
clinical covariates, PME was not associated with worse neuropsychological test
scores at age 10 or autistic traits at 19-20."
https://pubmed.ncbi.nlm.nih.gov/37283466/ [2713]
NIJZ (2017) had some information about ADHD, but about
ADHD in Slovenia not so much.
"Various epidemiological studies have found that for
the primary school population, the prevalence ranges from 2.4% to 19.8%. A
recent meta-study on 175 different prevalence studies carried out over the last
36 years estimated the prevalence of hyperkinetic disorder at 7.2%. The disorder
is more common in boys, with a sex ratio of 3-4:1. It should be pointed out that
the impact of sex is not yet fully understood and that the sex ratio almost
evens out in adulthood.
"In Slovenia, there are no data on prevalence and
incidence, but there are data on visits to GPs or specialist outpatient clinics
for hyperkinetic disorder, where an increase in the number of visits can be
observed in recent years."
The causes:
"The cause of the disorder is not yet fully understood
and is likely to be a combination of environmental and genetic risk factors, the
latter of which play a primary role:
"Genetic factors (family history of the disorder).
Neurophysiological factors (differences in the frontal
regions of the brain - reduced volume of the prefrontal cortex and reduced
thickness of the anterior cingulate cortex, as well as cortical thinning in both
upper frontal regions of the brain).
"Neurochemical factors (neurochemical peculiarities -
in particular in the action of neurotransmitters that affect executive functions
and cause excessive activity, distractibility or impulsivity - excessive
noradrenaline activity, dopamine deficiency).
"Psychosocial factors (stressful psychiatric events,
anxiety-provoking factors, the child's temperament, emotional deprivation and
society's demands that behaviour be adapted to the environment)."
https://web.archive.org/web/20220519122733/https://www.nijz.si/sl/bi-prepoznali-hiperkineticno-motnjo
[1883]
Dopamine was first identified in 1910 as an
intermediary in the synthesis of adrenaline and noradrenaline.
https://physoc.onlinelibrary.wiley.com/doi/epdf/10.1113/jphysiol.1910.sp001392
[2200]
It was not until 1957 that it was found in the brains
of several species including humans, and in the following years:
"...studies on the mechanisms of the first-generation
of antipsychiatric drugs, pioneered by Arvid Carlsson, identified dopamine as a
neurotransmitter playing a role in motor and mental functions. In parallel, Oleh
Hornykiewicz and others found that dopamine plays a critical role in movement
regulation in Parkinson disease. Finally, while studying the mechanisms of slow
neurotransmission, Paul Greengard and others revealed how dopamine acts on
dopamine receptors, activates downstream signalling pathways, and modulates
neuronal activity and synaptic plasticity." [2199]
So here, for instance, we see work on the effects of
drugs on adrenalin and noradrenaline steaming ahead in 1954, with not a mention
of dopamine, which had been so named in 1952. It is there, though, lurking in
the "total amines", for example in Table 15.
More than a quarter of a century after the Opium
Conference, dopamine's roles in motor control, modulation of behavior and
cognition, motivation and reward, inhibition of prolactin production, sleep,
dreaming, mood, attention, working memory, and learning was as yet unknown. [2205]
But in Table 9 we can see that cats on caffeine had
hypothalamic noradrenaline 109% of the control's. In cats on insulin it was
66.6% of the normal value, while morphine hydrochloride produced 56.6% of the
control or lower.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1366219/pdf/jphysiol01416-0041.pdf
[2202]
In 1957 came the recognition of dopamine as a
neurotransmitter:
https://www.nature.com/articles/1801200a0.pdf [2201].
As of 2022, dopamine unknowns included the
"...precise mechanisms and locations along the axons
and dendrites that dopamine is released, the structure and organization of
dopamine receptors, dopaminergic neuron subpopulations, their projections and
regulations, the role of glial cells in shaping dopamine functions, the patterns
of dopamine release at a single synapse, and across large brain areas, and the
time scale of dopamine modulation on intrinsic neuronal excitability and
synaptic plasticity."
https://www.frontiersin.org/research-topics/27370/brain-dopaminergic-mechanisms
[2199]
Dopamine theory remains restless. In a 2023 paper in
Nature Neuroscience, as explained in Wired's article "Everyone Was Wrong About
Antipsychotics":
"[Northwestern University neuroscientist Jones] Parker
shows that an assumption about antipsychotics that’s almost as old as the drugs
themselves is …. well, wrong.
"Neuroscientists have long thought that antipsychotics
dampen extreme dopamine transmission by sticking to receptors in a type of cell
called spiny projection neurons, or SPNs. The drugs basically box out the
dopamine at receptor proteins called D1 or D2 (where 'D' stands for dopamine).
Each of the spiny neurons sport either D1 or D2—they’re genetically distinct.
Experiments on calf brain extracts in the 1970s showed that the most powerful
antipsychotics are the ones that cling strongly to the D2 SPNs in particular, so
decades worth of antipsychotics were designed and refined with D2 in mind.
"But when Parker’s team probed how four antipsychotics
affect D1, D2, and mouse behavior, they found that the most drug interaction is
actually happening at D1 neurons."
By using 2g microscopes to peer into living mouse
brains via a tiny endoscope, Parker was able to study a model of amphetamine
psychosis and the effect of haloperidol, olanzapine, clozapine and a failed drug
candidate MP-10.
"The notion that D1 receptors may be a more important
target upends decades of research in a $15 billion market for drugs that are
famously erratic. Antipsychotics don’t work for about 30 percent of people who
try them. They’re plagued by side effects, from extreme lethargy to unwanted
facial movements, and rarely address the cognitive symptoms of psychosis, like
social withdrawal and poor working memory."
Parker's next plan is to see what happens with D1
partial agonists.
"The drugs compensate for high dopamine and low
dopamine. It’s a different approach than just blocking dopamine altogether, and
Parker hopes his new results bode well for D1 partial agonists in particular.
That’s because despite having more dopamine in their striatum, people with
schizophrenia actually have lower dopamine levels in their cortex, a feature
that neuroscientists think contributes to social withdrawal and forgetfulness.
'Such a drug could be both antipsychotic and cognition-promoting,' Parker says.
His lab has begun testing candidates."
https://www.wired.com/story/everyone-was-wrong-about-antipsychotics/?utm_medium=social&utm_source=twitter&utm_brand=wired-science&mbid=social_tw_sci&utm_social-type=owned
[2875]
https://www.nature.com/articles/s41593-023-01390-9 [2876]
Were it not for those pesky patents, Parker would not
need to look very far, as describing their work which "mark[ed] the first
demonstration of partial agonist/antagonist effects of THC in vivo" in 2012,
Paronis et al, over at Northeastern University in Boston, Mass., explain:
"The designation of a drug as a full or a partial
agonist is always related to the effects of other drugs in that pharmacological
class on the variable being measured. Thus, although we find that THC is a
partial agonist in producing hypothermia in mice, it must still be considered a
full agonist under conditions in which it produces the maximum possible effect,
including antinociception, decreased locomotor activity, and THC discrimination
(Compton et al., 1992; Fan et al., 1994; McMahon and Koek, 2007; Ginsburg et
al., 2012). Some studies have used the strategy of decreasing the number of
available receptors to rank the relative efficacy of opioid drugs that have full
agonist effects in vivo (Adams et al., 1990; Paronis and Holtzman, 1992). A
similar approach has been used to define THC as a partial agonist indirectly,
insofar as it shows greater tolerance than other cannabinoid agonists in vivo
(Hruba et al., 2012). Our results extend these findings by indicating that acute
administration of THC has partial agonist and antagonist effects in otherwise
drug-naive animals. Insofar as the apparent partial or full agonist effects of
drugs reflect their intrinsic properties, it seems likely that THC in vivo has
lower efficacy than AM2389 and, as has been shown in vitro, other cannabinoid
agonists."
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3697741/ [2877]
In 2023 an international collaboration
"...compared the shared genetic risk and biological
foundations of neurological and mental illnesses using roughly one million cases
from genome-wide association studies (GWAS)."
"Psychiatric disorders were more polygenic than
neurological disorders, with pediatric-onset disorders having the highest single
nucleotide polymorphism (SNP) heritability. The finding supported the hypothesis
that multiple causal pathways may converge on the same mental illness while
fewer causal pathways may underlie neurological disorders.
"The estimated polygenicity for psychiatric diseases
and COG [general cognitive ability] was greater than that for neurological
diseases, somatic disorders, cortical imaging evaluations, and height. Most
polygenic phenotypes had low discoverability, indicative of a higher proportion
of trait-affecting variants with smaller effect sizes.
"The study found that 40 of 45 genetic correlations
among psychiatric disorders and 12 of 45 correlations among neurological
disorders reached significance."
https://www.news-medical.net/news/20230801/Research-reveals-surprising-genetic-overlap-between-neurological-and-psychiatric-disorders.aspx
[2878]
https://www.medrxiv.org/content/medrxiv/early/2023/07/23/2023.07.21.23292993.full.pdf
[2879]
A 2020 review of ADHD studies points to a strong
genetic link.
"The formal heritability of ADHD is about 80%."
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7046577/ [1890]
Do you think ADHD [known as HKM in Slovene] is
associated with criminal behaviour?
PsychCentral.com has some ideas about why people with
ADHD might lie:
"Impulsivity often plays a role in why people with
ADHD lie.
"Sam Goldstein, PhD, a licensed psychologist in Utah,
explains people with ADHD have a tendency to act without thinking first while
under stress (impulsive behavior).
“'This alone [may] lead to an increased probability
that an impulsive person may lie to avoid responsibility or manipulate others to
achieve a goal,' Goldstein says.
"Still, he clarifies that 'there’s limited, if any,
scientific evidence that ADHD itself drives deceitful behavior. However,
combined with other personality and mental health challenges may lead to an
increased risk of lying.'
"Some people with ADHD may develop a habit of lying,
which, for some, could be a form of compulsive lying.
"Although lying can be a disruptive behavior, white
lies can often be harmless in nature. For example, difficulty staying focused
during a conversation can lead to someone lying to pretend like they were
listening to not hurt someone’s feelings.
"People with ADHD with a poor memory might also forget
something that happened, then say it didn’t when it actually did. To the other
person in the conversation, this may appear as lying.
"Some other reasons why adults or kids with ADHD may
lie may include:
"covering up an impulsive behavior that resulted in an
unwanted consequence forgetting what happened and lying to pretend like they
remember responding impulsively with a lie due to hyperactivity hiding a lack of
understanding of something with a lie wrongly answering questions they didn’t
listen to because they were distracted telling white lies out of difficulty
expressing themselves impulsively making promises they can’t keep Challenges
with executive functions can also make it harder for people with ADHD to process
information or speak and listen clearly. This could lead to miscommunications,
which may wrongly be considered lies."
https://psychcentral.com/adhd/adhd-and-lying#explanation [1882]
According to "The Relationship between Adult Symptoms
of Attention-Deficit/Hyperactivity Disorder and Criminogenic Cognitions" (2019)
by Englehardt et al
"The relationship between ADHD—in particular
hyperactivity—and criminal behavior is well documented."
and
"The first multiple regression examined whether the
factor-derived subscales predicted total criminogenic cognitions. The overall
model was significant F(4,187) = 52.13, p < 0.001. The R2 was 0.53, and age,
gender, inattention/memory problems, and impulsivity/emotional lability were all
retained as predictors (see Table 4). As predicted, higher age and being female
were negatively related to criminogenic cognitions, and the factor-derived
subscales were positively related to criminogenic cognitions. However, contrary
to expectations, inattention/memory problems was more strongly associated with
criminogenic cognitions than was impulsivity/emotional lability."
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6627881/ [1879]
120 years after Dr Still's lecture, instead of
measuring big heads, we now measure small brains.
According to "Subcortical brain volume differences in
participants with attention deficit hyperactivity disorder in children and
adults: a cross-sectional mega-analysis" by Hoogman et al, and published in the
Lancet (2017):
"Our sample comprised 1713 participants with ADHD and
1529 controls from 23 sites with a median age of 14 years (range 4–63 years).
The volumes of the accumbens (Cohen's d=−0·15), amygdala (d=−0·19), caudate
(d=−0·11), hippocampus (d=−0·11), putamen (d=−0·14), and intracranial volume
(d=−0·10) were smaller in individuals with ADHD compared with controls in the
mega-analysis. There was no difference in volume size in the pallidum (p=0·95)
and thalamus (p=0·39) between people with ADHD and controls. Exploratory
lifespan modelling suggested a delay of maturation and a delay of degeneration,
as effect sizes were highest in most subgroups of children (<15 years) versus
adults (>21 years): in the accumbens (Cohen's d=−0·19 vs −0·10), amygdala
(d=−0·18 vs −0·14), caudate (d=−0·13 vs −0·07), hippocampus (d=−0·12 vs −0·06),
putamen (d=−0·18 vs −0·08), and intracranial volume (d=−0·14 vs 0·01). There was
no difference between children and adults for the pallidum (p=0·79) or thalamus
(p=0·89). Case-control differences in adults were non-significant (all p>0·03).
Psychostimulant medication use (all p>0·15) or symptom scores (all p>0·02) did
not influence results, nor did the presence of comorbid psychiatric disorders
(all p>0·5)."
Their interpretation of these and other results:
"With the largest dataset to date, we add new
knowledge about bilateral amygdala, accumbens, and hippocampus reductions in
ADHD. We extend the brain maturation delay theory for ADHD to include
subcortical structures and refute medication effects on brain volume suggested
by earlier meta-analyses. Lifespan analyses suggest that, in the absence of well
powered longitudinal studies, the ENIGMA cross-sectional sample across six
decades of ages provides a means to generate hypotheses about lifespan
trajectories in brain phenotypes."
https://www.thelancet.com/journals/lanpsy/article/PIIS2215-0366(17)30049-4/fulltext
[1880]
What do the diagnosed ADHD cases, with their
apparently permanently misdeveloped brains, think about the utility of cannabis?
In "'I Use Weed for My ADHD': A Qualitative Analysis
of Online Forum Discussions on Cannabis Use and ADHD" Mitchell et al in North
Carolina examined 268 online forum threads. 20% were then randomly selected.
This 20% was then whittled down for various reasons, leaving 46 threads
containing 880 individual posts of which 401
"Twenty-five (25%) percent of individual posts
indicated that cannabis is therapeutic for ADHD, as opposed to 8% that it is
harmful, 5% that it is both therapeutic and harmful, and 2% that it has no
effect on ADHD. This pattern was generally consistent when the year of each post
was considered. The greater endorsement of therapeutic versus harmful effects of
cannabis did not generalize to mood, other (non-ADHD) psychiatric conditions, or
overall domains of daily life. Additional themes emerged (e.g., cannabis being
considered sanctioned by healthcare providers)."
Co-author Dr Kollins
"...has received research support and/or consulting
fees from the following: Akili Interactive, Alcobra, Arbor, Atentiv, Ironshore,
Neos, NIH, Neurovance, Purdue, Rhodes, Shire, Sunovion, and Tris in the past 2
years. This does not alter the authors’ adherence to PLOS ONE policies on
sharing data and materials. None of the other authors have any additional
declarations."
Here comes the "cannabis use disorder"...
"In the largest meta-analysis to date examining the
prospective association of ADHD with cannabis use, ADHD youth were nearly three
times as likely to report cannabis use in later life compared to non-ADHD youth;
and ADHD children were more than 1.5 times as likely to be subsequently
diagnosed with a CUD."
Well of course they are, because they are at the very
least 1.5 times more likely to come into contact with social workers who believe
in "cannabis use disorder". They are more likely to be in the justice or mental
health systems which take a criminal rather than a health-based perspective. All
these people would be very disappointed if cannabis turned out to be a net
positive. Their ignorance is motivated. This study does not consider these
effects on the prevalence - it's sole purpose is to analyse the fora.
The authors find
"...that at least three times as many comments
advocated for therapeutic effects of cannabis on ADHD compared to comments that
cannabis is harmful, both therapeutic and harmful, or has no effect on ADHD."
The authors do not mention placebo effect
specifically, but do admit:
"...no inferences can be drawn about the prevalence of
perceptions regarding the effects of cannabis on ADHD in patients with the
disorder—that was beyond the scope of the present study (i.e., to assess the
content of online data referring to cannabis and ADHD in forums)."
https://journals.plos.org/plosone/article/file?id=10.1371/journal.pone.0156614&type=printable
[1181]
Perhaps the reason cannabis and ADHD is not such a
popular area for hard science lies, again, in the desires of the competitors -
makers of Ritalin and a panoply of other ADHD drugs. A study of patients with
medical cannabis and ADHD diagnoses, 70% with other mental health conditions,
concluded:
"Although MC is not directly indicated for ADHD, low
ADHD symptom frequency and ADHD medication-sparing effects were found to be
associated with MC treatment. In addition, high dosage of CBN was associated
with lower ASRS [ADHD self-report scale], hinting at a possible combination
effect in whole-plant MC treatment. Nevertheless, although we found the
abovementioned association with CBN, it is minorly expressed in most MC
cultivars, thus, we assume that other phyto-cannabinoids might be more essential
for the effect on ADHD patients."
https://www.rmmj.org.il/userimages/1036/1/PublishFiles/1038Article.pdf
[1884]
Aleksi Hupli of the Tampere University in Finland
presents a case report in which an ADHD patient had gastric problems with
Ritalin and alcohol, and having heard about a delta-9 THC product Bedrocan was
able to move the mountain in just six months:
"After receiving this confirmation that the legal
framework supported his right to access cannabinoids, the patient began to
formally seek Bedrocan® as a substitute medication for methylphenidate. It was
hoped that cannabinoids would offer equivalent or better efficacy with more
tolerable adverse effects. After failing to find a Finnish psychiatrist or
neurologist with sufficient medical knowledge of CT, the patient exercised his
right to patient self-determination and finally, in June 2010, visited the
prescribing physician behind the small European ADHD study in Germany.
Afterwards, the patient returned to Finland with prescriptions for standardized
Bedrocan® and Bediol® medicinal cannabis products.
"Upon arrival to Finland, the next challenge for the
patient was to find a suitable Finnish physician to validate the prescriptions
for the cannabinoid treatment model. It took him until October 2010 − a period
of almost 4 months − to find a suitably qualified neurologist who was prepared
to endorse the treatment model. At that time, the patient presented the
prescribing neurologist with a challenge: no Finnish neurologist or psychiatrist
had previously substituted Bedrocan® for short-acting methylphenidate as a
pharmacological intervention for a neuropsychiatric medical condition. Clinical
guidelines for adult ADHD were only introduced in Finland in 2017, updating
pediatric treatment guidelines published in 2007, which were updated for
adolescents in 2013. These guidelines mention no possibility of CT for either
adult, adolescent, or pediatric ADHD. However, the Bedrocan® application was
submitted to Fimea in late November 2010 and approved by the end of December
2010."
Some other case reports are also presented.
https://pmc.ncbi.nlm.nih.gov/articles/PMC8489316/ [3935]
In search of mechanisms which might explain the
efficacy of cannabis for some with ADHD, NIH neurophysiologists Lupica et al in
"Marijuana and cannabinoid regulation of brain reward circuits" (2009) explain:
"Distinct physiological roles in which
endocannabinoids act as ‘retrograde messengers’ have been described in several
brain regions, including the NAc and VTA (Robbe et al., 2002; Melis et al.,
2004). In this capacity, endocannabinoids that are released from postsynaptic
neurons upon depolarization activate presynaptic CB1 receptors and inhibit
neurotransmitter release. This suggests that the endocannabinoid system may play
additional important roles in the regulation of ongoing synaptic brain function
(Alger, 2002; Wilson & Nicoll, 2002)."
The mechanisms elucidated by 2009 were:
"First, the ability of systemic cannabinoids to
increase extracellular DA concentrations in the NAc is reversed by systemic and
intra-VTA opioid antagonist administration (Chen et al., 1990; Tanda et al.,
1997), but the increase in DA neuron-firing rates caused by Δ9-THC are not
(French, 1997). Second, the direct infusion of Δ9-THC into the VTA does not
increase DA accumulation in the NAc (Chen et al., 1993). Third, it has recently
been demonstrated that synthetic cannabinoid agonists and endocannabinoids,
acting in a retrograde manner, can also inhibit glutamate release onto neurons
in the VTA in vitro (Melis et al., 2004), which would tend to diminish the
excitatory input to DA neurons in the VTA and reduce the probability of bursting
(Johnson et al., 1992; Kitai et al., 1999). Finally, preliminary data from our
laboratory indicate that CB1 receptors are also located on GABAergic terminals
believed to originate from NAc medium spiny output neurons (Walaas & Fonnum,
1980; Heimer et al., 1991) that target GABAB receptors on DA neurons in the VTA
(Sugita et al., 1992), suggesting a second possible disinhibitory mechanism
(Riegel et al., 2003). This latter study, taken together with that of Szabo et
al. (2002), implies that cannabinoids acting at CB1 receptors can inhibit the
release of GABA in the VTA that is derived from both intrinsic and extrinsic
sources, and further that the inputs from the NAc to the VTA may represent a
critical pathway for the expression of cannabinoid reward."
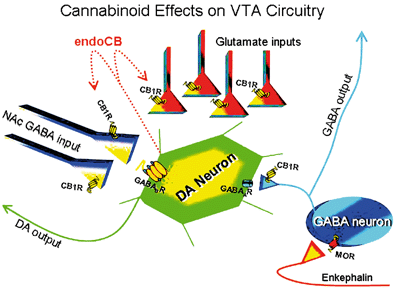
https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1038/sj.bjp.0705931
[1885]
By 2009 Albayram et al felt able to declare with certainty that
"Mice lacking the Cnr1 gene (Cnr1−/−), which encodes the cannabinoid receptor 1
(CB1), showed an accelerated age-dependent deficit in spatial learning
accompanied by a loss of principal neurons in the hippocampus....The ongoing
process of pyramidal cell degeneration and neuroinflammation can exacerbate each
other and both contribute to the cognitive deficits. Deletion of CB1 receptors
from the forebrain GABAergic, but not from the glutamatergic neurons, led to a
similar neuronal loss and increased neuroinflammation in the hippocampus as
observed in animals lacking CB1 receptors in all cells. Our results suggest that
CB1 receptor activity on hippocampal GABAergic neurons protects against
age-dependent cognitive decline by reducing pyramidal cell degeneration and
neuroinflammation."
And that:
"During aging, an increase in the expression levels of proinflammatory cytokines
takes place in the brain. We detected a significant increase in the expression
of IL-6 in 12-moold Cnr1−/− mice, whereas the expression of IL-1β, IL-6, or TNF
did not differ between 2-mo-old and 12-mo-old wild-type animals, in accordance
with previous reports. Changes in cell morphology and expression of surface
proteins and inflammatory cytokines have different dynamics and onsets.
Elevation of IL-6 levels has consistently been related to aging, and high levels
of IL-6 are associated with an increased risk of cognitive decline. The fact
that IL-6 but not IL-1β or TNF expression is increased suggests that the
increase in IL-6 expression is one of the first steps in the gradual activation
of microglial cells."
https://www.pnas.org/doi/full/10.1073/pnas.1016442108 [5404]
Obviously, back in 2009, these authors were stuck in an anti-cannabis mindset.
This has modified over the years. In 2022, to gauge
attitudes of perceived risk, Jack T Waddell of Arizona State University looked
at the trend:
"Public access data from the National Study on Drug
Use and Health from 2002 to 2019 were used (N = 1,005,421). Structural Equation
Models tested whether study year (linear trend), was associated with alcohol-
and cannabis-related risk perceptions (correlated outcomes), and whether age
(adolescence [12-17], emerging adulthood [18-25], adulthood [26-35], middle
adulthood [36-49], and older adulthood [50+]) moderated time trends. Sex,
race/ethnicity, and use frequency were covaried.
"Results: The linear trend of study year was
associated with decreased cannabis-related risk perceptions (p < .001). There
was also a significant interaction of age by study year for cannabis-related
risk perceptions, such that adults, emerging adults, and middle adults had the
largest decrease in attitudes over time. For alcohol-related risk perceptions,
the linear trend of study year was significantly associated with increased risk
perceptions (p = .001), but the interaction of time by age was non-significant;
alcohol-related effects were extremely small (b < 0.01)."
Mysteriously, Waddell concludes:
"Findings underscore the importance of targeting
permissive cannabis-related attitudes via prevention efforts."
But he doesn't say why all these people's perceptions
are wrong, or why he thinks his perception is better than all of theirs, or why
he thinks 18-49 year olds are particularly wrong.
https://pubmed.ncbi.nlm.nih.gov/34461500/ [1886]
Another study of risk perception around cannabis
enrolled 18,794 adults age ≥65 years participating in the 2015–2019 National
Survey on Drug Use and Health, a cross-sectional nationally representative
survey of non-institutionalized individuals in the U.S.
"Between 2015 and 2019, perceived risk associated with
regular use decreased from 52.6% to 42.7%, an 18.8% decrease (p<0.001).
Decreases in perceived risk were detected in particular among those never
married (a 32.6% decrease), those who binge drink (a 31.3% decrease), use
tobacco (a 26.8% decrease), have kidney disease (a 32.1% decrease), asthma (a
31.7% decrease), heart disease (a 16.5% decrease), chronic obstructive pulmonary
disease (a 21.5% decrease), two or more chronic conditions (a 20.2% decrease),
and among those reporting past-year emergency department use (a 21.0% decrease)
(ps<0.05)."
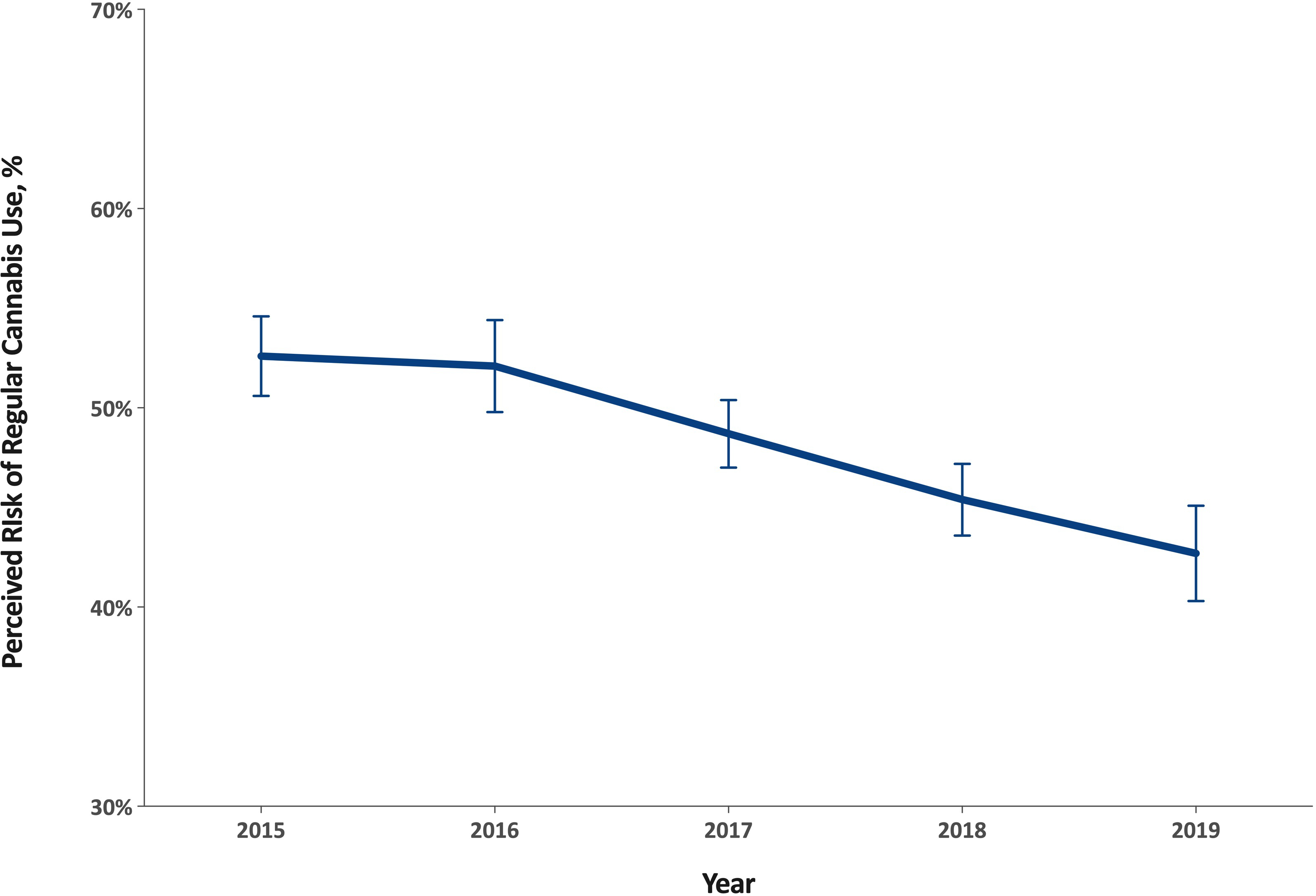
The authors note:
"The increase in interest for cannabis use as a
therapeutic drug for a variety of health conditions and its decrease in stigma
likely helps explain the drop in perceived risk among older adults."
and most tellingly about knowability as a problem for
the sheep-like mentality upon which prohibition depends:
"We also found a larger decrease in risk perception in
states where cannabis is legal compared to states where it is not."
A moment's reflection will reveal to the curious
onlooker that the legal status of cannabis and the sum of the physical harms and
benefits are independent variables: there is no mechanism whereby cannabis could
affect ADHD or insulin or PPAR-gamma or melanoma rates as a function of the law.
The only harm which could arise as a dependent variable of the legal status of
cannabis is harm caused by prohibition itself. Yet prohibition would like to
arrange matters such that it does not have to address health issues at all,
preferring to operate via innuendo and folk psychology
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8440375/ [1887]
So in relation to ADHD and cannabis, we have learned
at least three things: ADHD brain damage is irreparable, endocannabinoid
mechanisms are involved in the function of the VTA, and more patients think it
makes them think better than think the opposite.
In relation to cannabis safety generally, we have
learned along the way that large shifts in opinion towards cannabis are found in
people with no motivation to add to their health difficulties, and finally that
researchers in this area don't like this and are biased against cannabis
generally for no reason they care to explain.
"The global ADHD therapeutics market size is estimated
to be worth USD 29.56 billion in 2022 and USD 45.68 billion by 2027."
Besides COVID, which has boosted ADHD symptoms,
"The growing incidence of ADHD due to rough impact of
unstable lifestyles and additives in children’s diet across the world is
fundamentally driving the market growth as there is no treatment to for this
disorder."
https://www.marketdataforecast.com/market-reports/attention-deficit-hyperactivity-disorder-therapeutics-market
[1891]
My impression is Europe has nothing like the
cornucopia of drug choices available to American homo sapiens.
https://www.medicalnewstoday.com/articles/325201#medication-list [1892]
The prohibition narrative grinds on, as in Huang and
Lupica (2019) whose glass half-empty view is that
"...chronic Δ9-THC shifts the strength of
glutamatergic activation of NAc from cortical to sub-cortical limbic sites, and
we hypothesize that this contributes to deleterious effects of cannabis in
humans."
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7002212/ [1893]
Yet isn't this exactly what is claimed for Europe's
ADHD drug of choice Ritalin - methylphenidate hydrochloride?
https://pure.mpg.de/rest/items/item_1835660/component/file_1835659/content
[1894]
It turns out to be connected with the reason some
people like marijuana and some don't.
Rodents don't like THC, and why this should be is all
about this reward system.
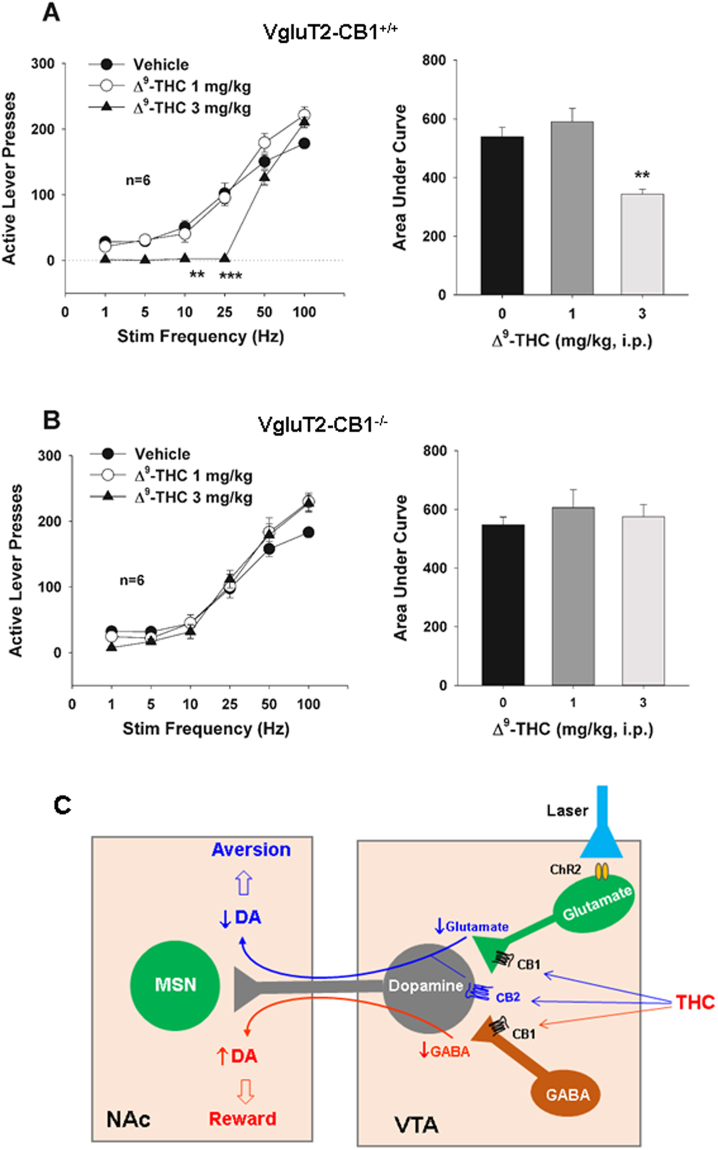
Thanks to Han et al (2017) who in "CB1 Receptor
Activation on VgluT2-Expressing Glutamatergic Neurons Underlies
Δ9-Tetrahydrocannabinol (Δ9-THC)-Induced Aversive Effects in Mice" attest that:
"Δ9-tetrahydrocannabinol (Δ9-THC), the major
psychoactive component of cannabis, produced dose-dependent conditioned place
aversion and a reduction in the above optical ICSS [intra cranial self
stimulation] in VgluT2-cre control mice, but not in VgluT2-CB1 −/− mice. These
findings suggest that activation of CB1Rs in VgluT2-expressing glutamate neurons
produces aversive effects that might explain why cannabinoid is not rewarding in
rodents and might also account for individual differences in the hedonic effects
of cannabis in humans."
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5614984/ [1896]
"The IRP–Beijing study, together with other evidence,
suggests that THC will be experienced as pleasurable or otherwise depending
largely on its net effect on two sets of neurons (see Figure 2). In addition to
glutamatergic neurons, the VTA is also home to neurons that release the
neurotransmitter gamma-aminobutyric acid (GABA). Previous studies have
demonstrated that these two types of neurons exert opposite effects on VTA
dopamine-releasing neurons. Whereas glutamatergic neurons stimulate the
dopaminergic neurons to release dopamine into the brain’s reward center,
GABA-ergic neurons inhibit them. Consequently, THC inhibition of VTA glutamate
neurons indirectly reduces dopamine activity in the reward center, leading to
aversion, and THC inhibition of GABA-ergic neurons increases dopamine activity,
producing euphoria.
"In the rodent VTA, the researchers note,
glutamatergic neurons produce more CB1 mRNA, and thus more CB1 receptors, than
do GABA-ergic neurons. Hence, when the rodent VTA is exposed to THC, the drug’s
inhibition of CB1 in glutamatergic neurons predominates, producing primarily
aversive effects. In the human VTA, in contrast, CB1 levels may be more similar
in glutamatergic and GABA-ergic neurons. As a result, when a person is exposed
to THC, the experience can be rewarding, aversive, or neutral."
https://nida.nih.gov/news-events/nida-notes/2018/03/why-marijuana-displeases
[1895]
An unpopular association was obtained by "Course of
Schizophrenia in Different Countries" (1987):

https://www.researchgate.net/publication/284638496_Course_of_Schizophrenia_in_Different_Countries_Some_Results_of_a_WHO_International_Comparative_5-Year_Follow-up_Study/link/588f03aca6fdcc8e63cbb90a/download
[4177]
...while Saha et al (2007) add:
"The prevalence of schizophrenia in migrants was
higher compared to native-born individuals: the migrant-to-native-born ratio
median (10%-90% quantile) was 1.8 (0.9-6.4). When sites were grouped by economic
status, prevalence estimates from "least developed" countries were significantly
lower than those from both "emerging" and "developed" sites (p = 0.04). Studies
that scored higher on a quality score had significantly higher prevalence
estimates (p = 0.02)."
https://journals.plos.org/plosmedicine/article?id=10.1371/journal.pmed.0020141
[4178]
And now...brain volumes and schizophrenia.
For the psychiatrists of Bergen, there was simply no
explanation. "Paradoxically," they say, "most neurocognitive studies on
schizophrenia have shown cannabis use to be a marker of superior performance on
neuropsychological tests."
Why is it paradoxical? Experimenter bias?
"A systematic literature review revealed better
cognitive functioning in cannabis-using compared to non-cannabis-using patients
in a majority of the reviewed 23 studies (Løberg and Hugdahl, 2009). This
pattern has been replicated by later studies (DeRosse et al., 2010;
Rodriguez-Sanchez et al., 2010), also including two meta-analyses (Rabin et al.,
2011; Yucel et al., 2012)."
It's all very worrying for the anti-cannabis dogma,
as:
"Studies comparing schizophrenia patients with and
without cannabis use by means of structural MRI and diffusion tensor imaging
(DTI) have shown more normalized (Dekker et al., 2010), more anomalous (Szeszko
et al., 2007; Bangalore et al., 2008; Rais et al., 2008; Ashtari et al., 2011;
Ho et al., 2011; James et al., 2011; Solowij et al., 2011), and equivalent
(Block et al., 2000; Cahn et al., 2004; Wobrock et al., 2009; Cohen et al.,
2012) brain anatomy in the cannabis group, thus making firm conclusions
difficult also when it comes to structural imaging."
In their own test on 26 schizophrenics with and
without previous cannabis use (but not current use), Løberg et al found
"...the Can+ group showed increased activation in the
task-present condition and decreased activation in the default mode network in
the absence of the task as compared to the Can− group."
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3483569/ [1923]
Cannabis schizophrenics had more insight and fewer
abusive or accusatory hallucinations:
"We used a case register that contained 757 cases of
first onset schizophrenia, 182 (24%) of whom had used cannabis in the year prior
to first presentation, 552 (73%) had not and 3% had missing data. We completed
the OPCRIT [Operational Criteria Checklist for Psychotic Illness] checklist on
all patients and investigated differences in the proportion of people with
distractibility, bizarre behaviour, positive formal thought disorder, delusions
of reference, well organised delusions, any first rank symptom, persecutory
delusions, abusive/accusatory hallucinations, blunted affect, negative thought
disorder, any negative symptoms (catatonia, blunted affect, negative thought
disorder, or deterioration), lack of insight, suicidal ideation and a positive
family history of schizophrenia, using chi square tests. Logistic regression
modelling was then used to determine whether prior cannabis use affected the
presence of the characteristics after controlling for age, sex and ethnicity.
"There was no statistically significant effect of
cannabis use on the presence of any of the above. There remained however a
non-significant trend towards more insight (OR 0.65 p = 0.055 for 'loss of
insight') and a finding of fewer abusive or accusatory hallucinations (OR 0.65 p
= 0.049) of borderline significance amongst the cannabis users. These were in
the hypothesised direction. There was no evidence of fewer negative symptoms or
greater family history amongst cannabis users."
https://www.sciencedirect.com/science/article/abs/pii/S0920996407001508?via%3Dihub
[1924]
Ibarra-Lecue et al (2021) believe:
"The most accepted theory is that daily use of
highpotency varieties of cannabis may trigger the onset of schizophrenia in
vulnerable individuals."
So their own findings have a special way of describing
unwanted results:
"The aim of the present study was to evaluate 5-HT2AR
protein expression and the Akt functional status in platelet homogenates of
subjects diagnosed with schizophrenia, cannabis use disorder, or both
conditions, compared with age- and sex-matched control subjects. Additionally,
endocannabinoids and pro-inflammatory interleukin-6 (IL-6) levels were also
measured in the plasma of these subjects. Results showed that both platelet
5-HT2AR and the active phospho (Ser473)Akt protein expression were significantly
increased in schizophrenia subjects, whereas patients with a dual diagnosis of
schizophrenia and cannabis use disorder did not show significant changes.
Similarly, plasma concentrations of anandamide and other lipid mediators such as
PEA and DEA, as well as the pro-inflammatory IL-6, were significantly increased
in schizophrenia, but not in dual subjects."
The authors have explained their position about
cannabis woo woo. This is a woo-woo way of saying:
"Platelet 5-HT2AR, active phospho (Ser473)Akt protein
expression, plasma concentrations of anandamide, PEA, DEA, and IL-6 were
significantly increased in schizophrenia subjects, unless they used cannabis."
https://onlinelibrary.wiley.com/doi/pdf/10.1111/adb.13233 [1969]
Perhaps we should not be surprised about Akt, aso
known as protein kinase B, as Ozaita et al (2007) say:
"We report that THC acute administration (10 mg/kg,
i.p.) increases the phosphorylation of Akt in mouse hippocampus, striatum, and
cerebellum. This phosphorylation was mediated by CB1 receptors as it was blocked
by the selective CB1 antagonist rimonabant."
and
"In conclusion, the present results demonstrate for
the first time in vivo that an exogenous cannabinoid, such as THC, activates the
neural-protective PI3K/Akt pathway and negatively regulates GSK-3b activity in
the mouse brain. These findings highlight the existence of cannabinoid-induced
activation of survival signaling pathways in the brain, as previously reported
in in vitro models. These molecular events provide new insights for better
understand the specific mechanisms involved in the neuroprotective effects that
have been reported after the activation of CB1 receptors by cannabinoid
agonists."
and
"Several studies have shown that cannabinoids can
protect neural cells from different insults, such as glutamatergic
excitotoxicity, oxidative damage, traumatic injury, and ischemia (for review,
see Guzman 2005). Some of these effects are linked to the activation of the
PI3K/Akt pathway, which is closely involved in the survival signaling in many
cell types including neurons. Cannabinoids can activate PI3K/Akt pathway by
acting on both CB1 and CB2 receptors (Sanchez et al. 2003), although the
protective effects on primary astrocytes (Gomez Del Pulgar et al. 2002) and
oligodendrocytes (Molina-Holgado et al. 2002) have been reported to involve CB1
receptor. The stimulation of the PI3K/Akt pathway is also required for the
neuroprotective effects of the synthetic cannabinoid HU-210 in primary cortical
neurons (Molina-Holgado et al. 2005)."
and
"We found a close regulation of Akt and GSK-3
phosphorylation by THC in brain, acting on CB1 receptors, that could be related
to the neuroprotective effects induced by cannabinoids in insults such as
ischemia, glutamatergic excitotoxicity, mechanical trauma, and oxidative damage
through the modulation of these crucial components of the cell survival
pathway."
and
"Considerable evidence exists demonstrating that
cannabinoids play a role as neuroprotective agents by both receptordependent
(reducing Ca2+ conductances and excitability) and receptor-independent
mechanisms (anti-oxidative properties of cannabinoid compounds) (reviewed in
Sarne and Mechoulam 2005). The signaling events involved in this beneficial
action produced in vivo are largely unknown. PI3K/Akt pathway promotes cell
survival by both enhancing the expression of anti-apoptotic proteins and
inhibiting the activity of pro-apoptotic ones. Direct intracellular targets of
PI3K/Akt involved in the control of apoptosis include Bad, caspase 9,
transcription factors of the Forkhead family, and GSK-3b (reviewed in Brunet et
al. 2001). The ability of cannabinoids to activate the pro-survival PI3K/Akt
pathway has been reported in some in vitro studies and may account for their
protective role (Gomez Del Pulgar et al. 2002; Molina-Holgado et al. 2002,
2005). Nevertheless, the signaling events mediated by CB1-receptor stimulation
in vivo remains poorly understood. The results presented herein show that in
vivo acute THC administration in mice activated Akt by enhancing Ser473
phosphorylation in the hippocampus, cerebellum, striatum and, to a minor extend,
in the frontal cortex. This effect was common to all the brain areas tested,
supporting the idea that this signaling mechanism is closely related to the
activation of CB1 receptors in the brain. The activation of Akt was dose
dependent with a modest effect at 0.3 mg/kg of THC, reaching the maximum peak at
10 mg/kg. Therefore, the dose of 10 mg/kg was used to characterize this
signaling pathway in vivo."
https://onlinelibrary.wiley.com/doi/pdf/10.1111/j.1471-4159.2007.04642.x
[1970]
Peineau et al (2008) have a diagram
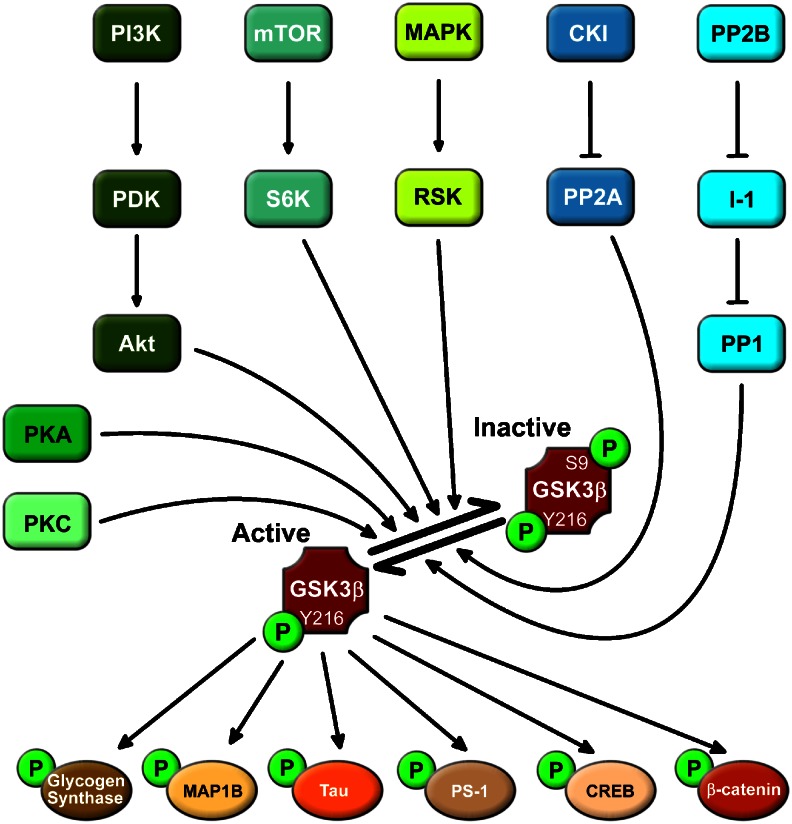
"Example of signalling pathways upstream and
downstream of GSK-3β. Under resting conditions, GSK-3β is basally activated by
phosphorylation at tyr216. Various ser/thr kinase cascades result in
phosphorylation of ser9 of GSK-3β, which results in inhibition of its activity.
Conversely, dephosphorylation of this residue results in disinhibtion of the
enzyme. GSK-3β phosphorylates a wide range of substrates. A selection of such
substrates that relate to neuronal function is shown. CREB, cAMP responsive
element-binding protein; CK1, casein kinase 1; I-1, inhibitor 1; MAP1B,
microtubule-associated protein 1B; MAPK, mitogen-activated protein kinase; mTOR,
mammalian target of rapamycin; PDK, phosphoinositide-dependent protein kinase;
PI3K, phosphatidylinositol 3-kinase; PP1, protein phosphatase 1; PP2A, protein
phosphatase 2A; PP2B, protein phosphatase 2B; PS-1, presenilin 1; RSK, p90
ribosomal S6 kinase; S6K, p70 ribosomal S6 kinase-1."
The authors have nothing to say about psychedelics,
but in discuss the link between GSK-3β and long term potentiation (LTP):
"Two independent studies have shown that following the
induction of LTP there is inhibition of GSK-3β (Hooper et al., 2007; Peineau et
al., 2007). This has been demonstrated following the induction of LTP in vivo in
both dentate gyrus and area CA1 in hippocampal slices (Figure 5a). The
inhibition of activity, assessed as an increase in phosphorylation of ser9, was
prominent 10–20 min after the induction of LTP and lasted for at least an hour.
This link between LTP and GSK-3β raises two questions. First, what influence
GSK-3β has on LTP and second, what role the LTP-induced regulation of GSK-3β
activity plays. With respect to the first issue, it was shown that in a
transgenic animal that overexpressed GSK-3β, there was a pronounced inhibition
of LTP (Figure 5b), which could account for the learning deficits observed in
these mice (Hernandez et al., 2002). This deficit was restored by treatment with
lithium, suggesting that it was the overexpression of GSK-3β that was
responsible for the effect rather than some developmental alteration (Hooper et
al., 2007). Could GSK-3β, given that it is ‘constitutively active', be providing
a tonic inhibition of LTP? In which case, GSK-3β inhibitors would be expected to
enhance LTP. Quantitative comparisons of the effects of a range of GSK-3β
inhibitors on LTP will be required to address this issue."
https://pmc.ncbi.nlm.nih.gov/articles/PMC2268071/ [4938]
In 2013 Koriyama et al demonstrated the therapeutic
role of GSK-3β inhibition in neurodegenerative diseases with an inflammatory
component:
"Activated microglial cells play an important role in
immune and inflammatory responses in CNS and play a role in neurodegenerative
diseases. We examined the effects of lipoic acid (LA) on inflammatory responses
of BV-2 microglial cells activated by lipopolysaccharide (LPS), and explored the
underlying mechanisms of action of LA. BV-2 cells treated with LPS showed an
up-regulation of mRNA of the pro-inflammatory molecules, inducible nitric oxide
synthase (iNOS). LA suppressed the expression of iNOS and furthermore,
LPS-induced production of nitrite. Moreover, LA suppressed the nuclear
translocation of RelA, a component of nuclear factor-kappa B (NF-κB) that
contains transcriptional activator domain for LPS. The mechanisms of LA-mediated
anti-inflammatory effects on microglia remain unknown, and we suggested an
involvement of Akt/glycogen synthase kinase-3β (GSK-3β) phosphorylation. The
results showed that inhibitor of phosphatidylinositol 3-kinase prevented
LA-mediated suppression of LPS induction of RelA and expression of iNOS.
Furthermore, these inflammatory actions were prevented by GSK-3β inhibitors."
https://www.sciencedirect.com/science/article/abs/pii/S0168010213001788?via%3Dihub
[4939]
According to Hans O Kalkman (2023) of the Psychiatric
University Hospital, University of Zurich:
"Risk factors for depression initiate an
infection-like inflammation in the brain that involves activation [of]
microglial Toll-like receptors and glycogen synthase kinase-3β (GSK3β). GSK3β
activity alters the balance between two competing transcription factors, the
pro-inflammatory/pro-oxidative transcription factor NFκB and the
neuroprotective, anti-inflammatory and anti-oxidative transcription factor NRF2.
The antidepressant activity of tricyclic antidepressants is assumed to involve
activation of GS-coupled microglial receptors, raising intracellular cAMP levels
and activation of protein kinase A (PKA). PKA and similar kinases inhibit the
enzyme activity of GSK3β. Experimental antidepressant principles, including
cannabinoid receptor-2 activation, opioid μ receptor agonists, 5HT2 agonists,
valproate, ketamine and electrical stimulation of the Vagus nerve, all activate
microglial pathways that result in GSK3β-inhibition."
https://www.mdpi.com/2227-9059/11/3/806 [4909]
In terms of convenience and safety, the Defendant
rules out all except the first of these as acceptable everyday experiences for
the purpose of inhibiting GSK3β - and adds a further example, curcumin, per
Bustanji et al (2008).
https://www.tandfonline.com/doi/10.1080/14756360802364377?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed
[4910]
This shows cannabis belongs to a group of
phytochemical sources capable of inhibiting glycogen synthase kinase-3β. Yet
curcumin is not listed as a drug with no medical purposes, is not banned and,
notably, does not make the consumer notably happier. No other reason for the
distinction can be ascertained.
Back on the statistical battlefield, and using as the
arbitrarily defined criterion of inclusion that "cannabis-users had to either
have a diagnosis of cannabis use disorder or use cannabis at least twice a
week", along with "psychopathology of individuals with schizophrenia spectrum
disorders assessed by the Positive and Negative Syndrome Scale (PANSS)",
"Association between cannabis use and symptom dimensions in schizophrenia
spectrum disorders: an individual participant data meta-analysis on 3053
individuals" by Argote et al trawled the entire history of publications on the
association up to September 2022.
Despite all this effort, and completely ignoring the
direction of association issue - i.e. whether schizophrenics were more likely to
become cannabis users rather than the reverse - the findings were unremarkable:
"Among the 1149 identified studies, 65 were eligible
and 21 datasets were shared, totaling 3677 IPD and 3053 complete cases. The
adjusted multivariate analysis revealed that relative to non-use, cannabis use
was associated with higher severity of positive dimension (3-factor: Adjusted
Mean Difference, aMD = 0.34, 95% Confidence Interval, CI = [0.03; 0.66];
5-factor: aMD = 0.38, 95% CI = [0.08; 0.63]), lower severity of negative
dimension (3-factor: aMD = −0.49, 95% CI [−0.90; −0.09]; 5-factor: aMD = −0.50,
95% CI = [−0.91; −0.08]), higher severity of excitement dimension (aMD = 0.16,
95% CI = [0.03; 0.28]). No association was found between cannabis use and
disorganization (aMD = −0.13, 95% CI = [−0.42; 0.17]) or depression (aMD =
−0.14, 95% CI = [−0.34; 0.06]). Interpretation No causal relationship can be
inferred from the current results. The findings could be in favor of both a
detrimental and beneficial effect of cannabis on positive and negative symptoms,
respectively. Longitudinal designs are needed to understand the role of cannabis
is this association. The reported effect sizes are small and CIs are wide, the
interpretation of findings should be taken with caution."
Despite their literary adhesion to the
cannabis-increases-schizophrenia hypothesis, the results if anything show an
opposite trend, such that the authors are forced to admit that despite their
hopes
"...the lower severity of negative symptoms for
cannabis-users cannot be ignored. The presented results support both the
selfmedication and toxicity hypotheses of cannabis use, with differential
effects on positive and negative symptoms."
https://www.thelancet.com/pdfs/journals/eclinm/PIIS2589-5370(23)00376-0.pdf
[4267]
Let's take a look at fold counts. This is a measure of
the overexpression or underexpression of some gene compared to a baseline. To
produce easier to handle numbers, fold count (FC) is expressed as a logarithm in
base 2, so you will see logFC.
"Fold change is the number of times a gene is
over-expressed (or under), compared to some baseline (your control, or the
reference gene, etc.). A sample could be 100X more expressed, or 1/100th the
expression of the baseline. Because this is hard to show in a graph, we plot in
log. It "flattens" the data out to make it more visible.
"Furthermore, because we tend to think of expression
in terms of copies of genes, or rather copies of copies of copies, we think of
it in terms of doubling which is why Log2 is frequently used to display the data
- you show the not the quantity, but the rounds of amplification of it. to give
better context between exponential differences in gene expression.
"In the instance of 'no difference' between a sample
and its baseline, or logFC = 0, the fold change, or ratio of a sample and
control is one, or one-to-one.
"If a sample is expressed twice as much as the control
(FC = 2), the logFC = 1; one doubling of the gene compared to baseline.
"So, to answer your question: if logFC = -0.5, then FC
= 2-0.5, or 0.7071, which means about 70% of the baseline, not 50%... 50%
reduction in expression would be a logFC of -1.
"if LogFC if 0.05, then your actual fold change is
1.0353... which is effectively 1, or rather, no significant change.
"To convert a logFC value, simply use it as the
exponent of two: 2logFC. In Excel, use the function "=2x". To convert a FC
value, take the log2. In Excel, use function: "=log(x,2). (where x = the cell
with your data)."
https://www.reddit.com/r/labrats/comments/7odtki/dumb_question_about_logfc/
[1897]
The relevance of this becomes clear when we look at
"THC exposure of human iPSC neurons impacts genes associated with
neuropsychiatric disorders" which the authors claim, in very controlled
language, to show
"significant alteration in THC-related genes
associated with autism and intellectual disability, suggesting shared molecular
pathways perturbed in neuropsychiatric disorders that are exacerbated by THC."
Reading the text you would hardly guess that not all
of these alterations are of the type "more gene alteration equals more
schizophrenia and more autism and more intellectual disability". Nothing could
be further from the facts, the reality is far more nuanced.
"There is a significant association between cannabis
use and schizophrenia in human subjects, however, whether this reflects patient
self-medication of prodromal symptoms or an environmental modulation of genetic
susceptibility remains an ongoing discussion. We recently reported molecular
abnormalities in schizophrenia patient hiPSC-derived neurons in response to
neural activity; here we describe a distinct overlap in hypo-excitability,
particularly in the glutamate system, between schizophrenia patient-derived
neurons and those treated with THC. THC exposure seems to deregulate glutamate
receptors and other genes involved in synaptic function. We observe significant
THC-dependent changes in postsynaptic density, ion channel and WNT
[Wingless/Int1 Trail] pathway genes, and epigenetic regulators; and molecular
connections to autism and intellectual disability. Although the molecular
mechanisms may not be precisely the same, the convergence of glutamatergic
hypo-function may partially explain the increased risk for psychiatric disorders
amongst those exposed to cannabis."
What do they mean by "not precisely the same"? It
means the molecular mechanisms are different.
"Relative to vehicle treatment, acute THC exposure
resulted in 497 genes significantly altered in hiPSC- derived neurons compared
to untreated controls, while chronic THC exposure perturbed 810 genes (Fig. 1a;
Supplementary Table S3; Supplementary Figure 1)."
Let's take a look at Supplementary Table S3
https://static-content.springer.com/esm/art%3A10.1038%2Fs41398-018-0137-3/MediaObjects/41398_2018_137_MOESM4_ESM.pdf
[1898]
We can see logFCs for the "perturbed" genes - genes
which are of course perturbed not only by THC but by all sorts of things - and
it is easily observed that some are numbers above 0 and some are below 0. The
Court will recall that a logFC of 1 represents a doubling of expression, a logFC
of -0.5 is about 70% of the control value.
If we look at the authors' Figure 2a we can see the
non-log fold changes are almost all positive - in this case remember the
no-change value is 1. We don't necessarily know whether more or less of some
activation is a good thing or a bad thing, or for whom.
The authors have selected HOMER1 for their example in
2b, which is the only lowered value among the postsynaptic density genes, for
"acute" THC, and one of only three out of 15 with a lowered fold count in the
"chronic" THC model.
2c shows postsynaptic density and ion channel genes
plotted by function but, again, there's nothing here showing anything more than
"perturbation" - we don't know if we want each of these perturbing, or in which
direction.
"Network analysis combining all THC-related genes from
acute and chronic THC treatment shows broad changes to fundamental cellular
functions such as RNA biology, chromatin regulation and development."
In Figure 3 the perturbed genes are counted by
association with THC and the three disorders. Again this says nothing about
positive or negative influences per se, and a certain degree of tunnel vision is
already developing.
"We noticed that many genes implicated in psychiatric
disease coincided with genes altered in response to THC treatments. In order to
calculate statistical relevance we needed to first update the numbers of genes
associated with these disorders and found genes related to autism spectrum
disorder (1037 genes), intellectual disability (2461 genes) and schizophrenia
(723 genes; see Supplementary Information ‘Generation of Gene Databases’ for
details; Supplementary Table S7). Included in our list of significantly altered
transcripts following THC exposure is a substantial number of genes linked to
autism (80 genes) and intellectual disability (167 genes), with fewer
overlapping with schizophrenia (Fig. 3a); autism and intellectual disability
associated genes are significant for both p-value and odds ratio using the
Fisher’s exact test (Fig. 3b). These data suggest that endogenous THC responsive
pathways include many psychiatric disease-associated genes and that changes in
these genes, either genetically or epigenetically, may contribute to
cannabis-related adverse reactions such as psychosis in some users."
But the "suggestion" is not a valid statement - or at
least non-neutral - as the outcomes have not been shown to be universally
"adverse" at all. Indeed, although for intellectual disability and autism the
odds ratio for this association - not the disease - are 1.7 and 1.9
respectively, the odds for an association with schizophrenia - not the disease -
are less than unity - 0.9.
The association is between genes connected with THC
good or bad and genes connected with schizophrenia good or bad. It tells us
nothing about THC and schizophrenia...
The well-known gene for dopamine metabolism COMT is
found in the autism and schizophrenia lists at Supplementary Table 7, but not
the intellectual disability list.
https://static-content.springer.com/esm/art%3A10.1038%2Fs41398-018-0137-3/MediaObjects/41398_2018_137_MOESM8_ESM.pdf
All the "THC-related pathways" are also
anandamide-related pathways. As early as 1998 Adams et al the Medical College of
Virginia wrote:
"Anandamide is the newly discovered endogenous
cannabinoid ligand that binds to brain cannabinoid receptors and shares most,
but not all, of the pharmacological properties of delta 9-THC. Therefore, this
study was undertaken to determine whether its interaction with the CB1 receptor
in brain was identical to that of delta 9-THC. Anandamide depressed spontaneous
activity and produced hypothermia, antinociception and immobility in mice after
i.v. administration. However, none of these effects was blocked by pretreatment
with the selective CB1 antagonist, SR 141716A. However, the metabolically stable
analog 2-methyl-2'-fluoroethylanandamide produced reductions in motor activity
and antinociception in mice, effects that were blocked by the antagonist. To
determine whether anandamide's receptor binding mimicked that of other
cannabinoids, an autoradiographic comparison of anandamide, SR 141716A and CP
55,940 competition for [3H]CP55,940 binding was conducted throughout rat brain.
The receptor affinities for all three compounds did not change according to
brain area. As expected, Bmax values differed dramatically among differ brain
areas. However, the Bmax values for each brain area were similar regardless of
the compound used for displacement. These data suggest that anandamide, SR
141716A and CP 55,940 compete for the same cannabinoid receptor throughout brain
despite SR 141716A's failure to block anandamide's pharmacological effects.
Although there is no question that anandamide binds to the cannabinoid receptor,
failure of SR 141716A to block its pharmacological effects in mice poses a
dilemma. The results presented herein raise the possibility that anandamide may
not be producing all of its effects by a direct interaction with the CB1
receptor."
https://pubmed.ncbi.nlm.nih.gov/9495885/ [1929]
As for the Jaccard Index
"The class also calculates the Jaccard index which
measures the similarity between two lists. The Jaccard index varies between 0
and 1, with 0 meaning there is no similarity between the two and 1 meaning the
two are identical."
The findings prove nothing for any individual case and
are more aimed at pharmaceutical research.
https://static-content.springer.com/esm/art%3A10.1038%2Fs41398-018-0137-3/MediaObjects/41398_2018_137_MOESM1_ESM.pdf
[1900]
Justin Jackson at medicalxpress.com writes:
"Debate continues regarding the nature of the
association between adolescent cannabis use and psychosis risk, with theories
including the contributing risk hypothesis, the shared vulnerability hypothesis,
and the self-medication hypothesis.
"In the contributing risk hypothesis, cannabis use
causes the emergence and progression of psychosis through disruption of the
neurodevelopmental processes during adolescence.
"According to the shared vulnerability hypothesis,
genetic, gestational, or environmental factors predispose individuals to both
cannabis use and psychosis. In this scenario, the likelihood of engaging in
cannabis use shares the same origin as the risk of psychosis spectrum symptoms.
"The self-medication hypothesis suggests that
individuals may turn to cannabis use as a means to alleviate distressing
symptoms associated with the psychosis spectrum.
"Previous research has provided evidence supporting
each of these models, but there is a lack of prospective longitudinal studies
focusing on early adolescence.
"In a study, 'Psychosis Spectrum Symptoms Before and
After Adolescent Cannabis Use Initiation,' published online in JAMA Psychiatry,
the researchers analyzed psychosis spectrum symptom trajectories before and
after cannabis initiation in 11,868 adolescents aged 9 to 10 years at baseline
using data from five waves over four years from the Adolescent Brain Cognitive
Development (ABCD) Study.
"Cannabis initiation did not consistently lead to an
increase in psychosis symptoms, providing no significant support for the
contributing risk hypothesis.
"Adolescents who used cannabis at any point during the
study period reported a greater number of psychosis spectrum symptoms and more
distress compared to those who never used cannabis, supporting the shared
vulnerability hypothesis.
"An increase in the number of psychosis spectrum
symptoms and associated distress leading up to cannabis initiation was observed
before cannabis use started, aligning well with the self-medication hypothesis.
"Based on the findings, the current research supports
the shared vulnerability and self-medication explanations for the associations
between cannabis use and psychosis risk."
https://medicalxpress.com/news/2024-11-psychosis-symptoms-adolescent-cannabis.html
[3709]
The study, "Psychosis Spectrum Symptoms Before and
After Adolescent Cannabis Use Initiation" by Osborne et al (2024) reports:
"Among the 11 858 participants at wave 1, the mean
(SD) age was 9.5 (0.5) years; 6182 (52%) participants were male. Consistent with
a shared vulnerability hypothesis, adolescents who used cannabis at any point
during the study period reported a greater number of psychosis spectrum symptoms
(B, 0.86; 95% CI, 0.68-1.04) and more distress (B, 1.17; 95% CI, 0.96-1.39) from
psychosis spectrum symptoms relative to those who never used cannabis.
Additionally, consistent with a self-medication hypothesis, the number of
psychosis spectrum symptoms (B, 0.16; 95% CI, 0.12-0.20) and distress (B, 0.23;
95% CI, 0.21-0.26) from psychosis spectrum symptoms increased in the time
leading up to cannabis initiation. We observed mixed evidence for an increase in
psychosis symptoms after cannabis initiation (ie, contributing risk
hypothesis)."
https://jamanetwork.com/journals/jamapsychiatry/article-abstract/2825423
[3710]
A case study demonstrates that:
"...in some cases people with ADHD may show unusual
effects after the consumption of THC. A 28-year-old male, who showed abnormal
behaviour and seemed to be significantly maladjusted and inattentive while
sober, appeared to be completely normal with a very high plasma level of THC.
Performance tests conducted with the test batteries ART2020 and TAP provided
average and partly above-average results in functions related to driving. Thus,
it has to be taken into account that in persons with ADHD THC may have atypical
and even performance-enhancing effects."
https://pubmed.ncbi.nlm.nih.gov/17879702/ [1903]
According to Aran et al at the Neuropediatric Unit,
Shaare Zedek Medical Center, Jerusalem, Israel (2019)
"Serum levels of the main endocannabinoids,
N-arachidonoylethanolamine (AEA or anandamide) and 2-arachidonoylglycerol
(2-AG), and their related endogenous compounds, arachidonic acid (AA),
N-palmitoylethanolamine (PEA), and N-oleoylethanolamine (OEA), were analyzed by
liquid chromatography/tandem mass spectrometry in 93 children with ASD
(age = 13.1 ± 4.1, range 6–21; 79% boys) and 93 age- and gender-matched
neurotypical children (age = 11.8 ± 4.3, range 5.5–21; 79% boys). Results were
associated with gender and use of medications, and were correlated with age,
BMI, and adaptive functioning of ASD participants as reflected by scores of
Autism Diagnostic Observation Schedule (ADOS-2), Vineland Adaptive Behavior
Scale-II (VABS-II), and Social Responsiveness Scale-II (SRS-2).
"Results
Children with ASD had lower levels (pmol/mL,
mean ± SEM) of AEA (0.722 ± 0.045 vs. 1.252 ± 0.072, P < 0.0001, effect size
0.91), OEA (17.3 ± 0.80 vs. 27.8 ± 1.44, P < 0.0001, effect size 0.94), and PEA
(4.93 ± 0.32 vs. 7.15 ± 0.37, P < 0.0001, effect size 0.65), but not AA and
2-AG. Serum levels of AEA, OEA, and PEA were not significantly associated or
correlated with age, gender, BMI, medications, and adaptive functioning of ASD
participants. In children with ASD, but not in the control group, younger age
and lower BMI tended to correlate with lower AEA levels. However, these
correlations were not statistically significant after a correction for multiple
comparisons."
In their conclusion, the authors of "Lower circulating
endocannabinoid levels in children with autism spectrum disorder" suggest using
these as a diagnostic:
"We found lower levels of the endocannabinoids AEA,
OEA, and PEA in serum samples of 93 children with ASD compared with samples of
matched neurotypical control group. These findings are in line with the results
of numerous former studies in animal models of ASD as well as an initial human
study that demonstrated lower endocannabinoid tone in ASD. Our findings suggest
the use of circulating AEA, OEA, and PEA as stratifying biomarkers of ASD and
future studies should assess the clinical significance of this stratification.
These markers can also be easily measured longitudinally in humans and in animal
models alike, and future studies should evaluate their potential to assist in
the monitoring of treatment response. Further studies are needed to determine
whether circulating endocannabinoid levels are also lower in infants and can
assist in pre-symptomatic diagnosis and if they reflect lower endocannabinoid
tone in the brain, as found in animal models of ASD."
https://molecularautism.biomedcentral.com/articles/10.1186/s13229-019-0256-6
[2051]
As Janna Champagne (2024) explains in "EndoCannabinoid
Deficiency and Autism" the general bias has been to ignore the unprofitable ECS
and gut-brain axis:
"Autism Spectrum Disorder (ASD) results from many
complex contributors creating pervasive imbalances. Research has theorized about
the involvement of dozens of genetic mutations for predisposing Autism, and
unfortunately the absence of targeted biomarkers spawned a high reliance on
behaviors to establish diagnostic criteria, as detailed in the Diagnostic &
Statistical Manual for Mental Disorders (DSM-V). This lends to classification of
ASD as a mental health disorder, and approaches for alleviating the underlying
physical imbalances are often overlooked or dismissed in favor of prescribing
mental health pharmaceuticals (Pietropaolo, 2021).
"However, as we know of other mental health disorders,
there are underlying physical imbalances contributing to Autism symptoms and
behaviors, primarily in the Gastrointestinal (GI) tract, plus the Immune &
Neurological systems. These imbalances may be identified and targeted through
biometric assessment, and interventions are proving beneficial for promoting
improved long- term outcomes. In recent years, many research studies reflect a
strong link between dysfunction of the EndoCannabinoid System (ECS) as a factor
in diagnosis of Autism (Pietropaolo, 2021).
"One major role of the ECS is creation of
EndoCannabinoids, from precursory intake of Omega fatty acids. These
EndoCannabinoids circulate throughout the body, similarly to the physiology of
the Endocrine system, producing substances and distributing them for interaction
with receptors located throughout the body. The ECS receptors are found in every
other system of the body, and the interactions at these sites promote systemic
homeostasis (Castillo, 2012)."
The problem is the alleged solution:
"The US Food & Drug Administration (FDA) approved
pharmaceuticals for addressing ASD symptoms are two anti-psychotics: Abilify and
Risperidone. Additionally, physicians often prescribe off-label use medications
as an attempt to de-escalate ASD behaviors, including Selective Serotonin
Reuptake Inhibitor (SSRI) anti-depressants, Benzodiazepine anxiolytics,
Ritalin/Adderall, and Anticonvulsants. These medications entail substantial risk
for side effects that may impair quality of life, including Extra-Pyramidal
Symptoms, seizures, mitochondrial damage, paradoxical effects, and male breast
development. In addition, use of these medications entails known risks of severe
adverse effects that may be life-threatening, such as Neuroleptic Malignant
Syndrome, Steven Johnson Syndrome, and suicidal ideation (Autism Research
Institute, 2024). Compared objectively with medical cannabis, and its mild or
pleasurable side effects, and no reports of substantiated harm, cannabis use
entails fewer risk factors (Goldstein, 2023)."
In her own results:
"Upon receiving treatment with medical cannabis, the
subjects with ASD were tested and salivary biomarkers were compared with the
prior levels from the ASD subjects, as well as the control group’s neurotypical
baseline levels. The result of this data compilation reflected that of the
sixty-five biomarkers included, twenty-three of the parameters shifted
positively towards neurotypical baseline (Siani-Rose, 2021).
"Additionally, the parent reporting reflected improved
symptom and behavior management after cannabis administration, compared with the
individual baseline. Overall, this evidence supports cannabis as a harm
reduction tool for addressing symptoms of ASD, strengthened by case studies,
patient outcomes, and this recent proof of improvement in ASD-applicable
biomarkers (Siani-Rose, 2021)."
https://www.researchgate.net/profile/Janna-Champagne/publication/388277723_EC_deficiency_and_Autism/links/67915d4495e02f182eae5dfb/EC-deficiency-and-Autism?_tp=eyJjb250ZXh0Ijp7ImZpcnN0UGFnZSI6InB1YmxpY2F0aW9uIiwicGFnZSI6InB1YmxpY2F0aW9uIn19
[4851]
At the time of the visit, did you know anything about
the Defendant's AEA, OEA or PEA levels?
More about PEA:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3744360/ [2053]
More about AEA, OEA, PEA, and SEA from Fezza et al
(2014)
"AEA belongs to a class of naturally occurring
molecules (NAEs) known for a long time. One of its members,
N-palmitoylethanolamine (PEA), was first reported almost 50 years ago in humans,
yet its physiological relevant remains under debate when the mechanism is other
than via CBRs.
"PEA and other NAEs share with true eCBs many
degradative mechanisms, and they potentiate the effect of eCBs at their receptor
targets by competitively inhibiting their hydrolysis, or by allosterically
modulating their receptor binding: the so-called 'entourage effect'. On this
basis, these substances are also known as 'eCBs-like' compounds (Table 1).
"Among the most studied eCBs-like compounds, the
anti-inflammatory agent PEA and the appetite-suppressor N-oleoylethanolamine
(OEA) can be listed (Table 1). Their biological activity often engages PPARα and
TRPV1 activation, although some of their actions are prevented by CB1
antagonists. Among the eCBs-like compounds OEA shows the highest affinity for
PPARα, and consistently some of its biological effects are absent in PPARα
deficient mice. Yet, the antinociceptive properties of OEA are exercised also
through a PPARα-independent mechanism.
"OEA, as well as PEA and 2-oleoylglycerol (2-OG)
(Table 1), can also activate GPR119, a GPCR expressed predominantly in human and
rat pancreas, suggesting that the effects of OEA on food intake may be mediated,
at least in part, via GPR119. Conversely, Lan and coworkers reported that the
hypophagic effect of OEA was preserved in Gpr119(-/-) mice. Not surprisingly,
there is also evidence that OEA (as well as PEA) can engage, even though at high
concentrations, additional receptors like GPR55.
"Another saturated NAE, N-stearoylethanolamine (SEA),
was reported to act as a cell growth controller and
anti-inflammatory/immunomodulatory agent, through yet unknown targets. SEA also
shows anorexic effects that are PPAR-independent and, together with PEA, plays
an antinociceptive role in humans.
"As reported above, also the endogenous levels of
these eCBs-like compounds (PEA, OEA and SEA) are affected by different dietary
regimens, with different hits in the brain compared to peripheral tissues."
[eCBs and eCBs-like compounds, molecular targets,
biosynthetic and catabolic enzymes 2054]
https://www.mdpi.com/1420-3049/19/11/17078 [2054]
Is alcohol connected with brain volumes?
In their narrative review of studies pertaining to the
assessment of CBD efficiency on drinking reduction, or on the improvement of any
aspect of alcohol-related toxicity in AUD, French researchers De Ternay et al
find:
"ARLD is a progressive alcohol-induced liver injury,
which starts with an increase in the amount of fat in the liver—a process called
steatosis—and continues into a progressive cell loss, fibrosis, and hepatic
insufficiency—a process called cirrhosis (O’Shea et al., 2010). ARLD may result
in severe liver failure, and represents a major risk factor for liver cancer.
Overall, alcohol-attributable liver damage is responsible for 493,300 deaths
every year, and 14,544,000 disability adjusted life years (DALYs), representing
0.9% of all global deaths and 0.6% of all global DALYs all over the world (Rehm
et al., 2013). In subjects with ARLD, preventing the transition from steatosis
to cirrhosis is a major treatment goal, and this usually requires to stop or to
dramatically reduce the average amount of consumed alcohol in the long term
(European Association for the Study of the Liver A et al., 2018). AUD also
affects the brain, through ARBI. Subjects with AUD display reduced gray matter
volumes and reduced cortical thickness, as well as increased ventricular
volumes, when compared to matching healthy controls (Bühler and Mann, 2011). The
most significant reductions in grey matter volumes are observed in the
corticostriatal–limbic circuits, including the insula, superior temporal gyrus,
dorso-lateral prefrontal cortex, anterior cingulate cortex, striatum, and
thalamus (Bühler and Mann, 2011). Cognitive functions associated with these
brain areas (e.g., executive functions, working memory, emotion recognition, or
long-term memory) are impaired in subjects with AUD (Stavro et al., 2013).
Generally, cognitive dysfunctions start to improve quickly after alcohol
withdrawal, but patients substantially recover only within the first weeks to
months of alcohol abstinence, and sometimes remain impaired (Stavro et al.,
2013; Schulte et al., 2014). Similarly, the recovery of structural brain
alterations can be highly variable depending on brain areas and individual
features (Durazzo et al., 2015; Zou et al., 2018). Overall, both ARLD and ARBI
involve alcohol-related inflammatory processes (Mandrekar and Ambade, 2014;
Neupane, 2016). Current medications for reducing alcohol drinking or supporting
alcohol abstinence in AUD subjects are still insufficiently effective at a
population level, and new therapeutic prospects are needed (Rolland et al.,
2016; Soyka and Müller, 2017). Moreover, no drug for reducing alcohol-related
harms, either on the brain or the liver, has ever been studied."
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6554654/ [1921]
Luckily we've been studying it for 12,000 years.
In "Regional Brain Volume Changes in Alcohol-dependent
Individuals during Short-term and Long-term Abstinence" Zou et al elaborate:
"Widespread brain atrophy in alcohol-dependent
individuals (ALC) has been consistently documented in pathological and magnetic
resonance imaging (MRI) studies. Longitudinal MRI studies have shown that the
regional brain volume losses in ALC are partially reversible during abstinence
from alcohol. The goal of this study was to determine volume reductions in
cortical and subcortical regions functionally important to substance use
behavior and their changes during short-term (1 week to 1 month) and long-term
abstinence (1 month to 7 months) from alcohol. The regions of interests (ROIs)
were: anterior cingulate cortex (ACC), dorsolateral prefrontal cortex (DLPFC),
orbitofrontal cortex (OFC), insula, amygdala, and hippocampus."
You can see the brain volumes increasing after one
week, one month, and 7 months of abstinence at their Figure 1
In their Table 2 you can see the amount of brain
involved. To begin with, all the drinkers' brains are smaller in volume than the
non-drinkers. For instance the dorsolateral prefrontal cortex grew back at a
quadratic rate of 28.0 mm3/month2.
In summary:
"The regional volumes in CON did not change
significantly over the 10-months scan interval, attesting to the stability of
our measurement paradigm over time. By contrast, in ALC, the volumes of DLPFC,
OFC, insula, and hippocampus increased over the TP1-TP2 (all p < 0.005) and
TP2-TP3 intervals (all p < 0.012). The ACC volume increased significantly only
over TP2-TP3 (p = 0.013), but the volume did not increase significantly over the
TP1-TP2 interval. The amygdala volume tended to increase only during the TP1-TP2
interval (p = 0.058) and remained practically unchanged after TP2. The linear
monthly volume change rates in the DLPFC, OFC, and insula were about 4 to 6
times higher during short-term than long-term abstinence (2.5 times for the
ACC), so that up to 50% of the observed volume increases in these ROIs over the
entire 7-months abstinence period occurred during the first month of abstinence.
Over the entire abstinence period, the DLPFC, OFC, and insula also showed
significant non-linear volume recovery trajectories, i.e., the quadratic monthly
volume change rates were significant (all p < 0.009) (see Table 2). The
significant quadratic rates of these three ROIs were all negative (i.e.,
frowning parabolas), indicating that the volumes of these three ROIs increased
faster during short-term abstinence than long-term abstinence."
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5984169/ [2868]
A 2021 UK Biobank study from Topiwala et al, was
"...one of the largest imaging investigations into the
impact of alcohol consumption on brain health to date. The very large sample
size provided great statistical power to detect associations across almost the
whole cortex, subcortical structures and cerebellum that have previously been
uncharacterized, as well as extensively explore interactions with clinical and
drinking behaviours. It used state-of-the-art neuroimaging and results were
stringently controlled for more potential confounders than ever before, as well
as multiple testing which increases confidence in the findings."
Which showed no lower threshold for alcohol damage in
terms of grey matter volume:
"Alcohol consumption was negatively linearly
associated with global brain grey matter volume (beta= -0.1, 95%CI= -0.11 to
-0.09, p<2x10-16). The association with alcohol was stronger than other
modifiable factor tested and robust to unobserved confounding. Widespread
negative associations were observed with white matter microstructure (beta=
-0.08, 95%CI= -0.09 to -0.06, p<2x10-16) and positive correlations with
functional connectivity. Higher blood pressure and body mass index increased
risk of alcohol-related harm (SBP*alcohol: beta= - 0.01, 95%CI = -0.02 to
-0.004, p=0.005; BMI*alcohol: beta= -0.01, 95%CI = -0.02 to -0.002, p=0.02).
Binging on alcohol had additive negative effects on brain structure on top of
the absolute volume consumed (daily compared to never binging: beta= -0.19,
95%CI= -0.30 to -0.08, p<0.01). No evidence was found for differential effects
of drinking wine, beer or spirits.
"Conclusions
No safe dose of alcohol for the brain was found.
Moderate consumption is associated with more widespread adverse effects on the
brain than previously recognised. Individuals who binge drink or with high blood
pressure and BMI may be more susceptible. Detrimental effects of drinking appear
to be greater than other modifiable factors. Current 'low risk' drinking
guidelines should be revisited to take account of brain effects."
https://www.medrxiv.org/content/10.1101/2021.05.10.21256931v1.full.pdf
[3542]
In 2022's "Association Between Brain Structure and
Alcohol Use Behaviors in Adults: A Mendelian Randomization and Multiomics Study"
Mavromatis et al set out in JAMA Psychiatry:
"To use mendelian randomization (MR) to identify
directional associations between brain structure and alcohol use and elucidate
the transcriptomic and cellular underpinnings of identified associations."
as
"Recently developed genomics methods, including latent
causal variable analysis and mendelian randomization (MR), facilitate the
identification of directional associations between genetically influenced
variables from population based observational data and have been underapplied to
questions regarding alcohol use and brain structure."
and
"The main bidirectional MR analyses were performed in
samples totaling 763 874 individuals, among whom more than 94% were of European
ancestry, 52% to 54% were female, and the mean cohort ages were 40 to 63 years.
Negative associations were identified between genetically predicted GCT and
binge drinking (β, −2.52; 95% CI, −4.13 to −0.91) and DPW (β, −0.88; 95% CI,
−1.37 to −0.40) at a false discovery rate (FDR) of 0.05. These associations
remained significant in multivariable MR models that accounted for
neuropsychiatric phenotypes, substance use, trauma, and neurodegeneration. TWAS
[transcriptome-wide association studies] of GCT [global cortical thickness] and
alcohol use behaviors identified 5 genes at the 17q21.31 locus oppositely
associated with GCT and binge drinking or DPW [drinks per week] (FDR = 0.05).
Cell-type enrichment analyses implicated glutamatergic cortical neurons in
alcohol use behaviors."
and
"Our large sample sizes (between 19 629 and 537 349
participants) increased statistical power relative to previous brain
structure–alcohol consumption studies. Our findings suggest that a
predisposition toward lower GCT may be associated with greater alcohol
consumption and binge drinking. Conversely, we failed to find strong evidence
that a genetic predisposition for alcohol associated with brain structure or its
longitudinal plasticity."
and
"Our investigation of the transcriptomic relationship
between GCT and alcohol use identified 5 protein coding genes oppositely
associated with GCT and alcohol use behavior: PLEKHM1, LRRC37A2, CRHR1,
ARHGAP27, and LRRC37A. These 5 genes could contribute to the negative
association between GCT and alcohol use. All 5 are located at 17q21.31. This
locus, characterized by extensive linkage disequilibrium, is the site of 2
haplotypes: the inverted H2 haplotype (found in approximately 20% of individuals
of European ancestry), and the H1 haplotype."
and
"Our cell-type analysis also found that excitatory
neurons may underlie GCT’s association with alcohol use. These data support the
notion that glutamatergic transmission plays an important role in alcohol
misuse. Interestingly, CRHR1 is expressed in glutamatergic, but not GABAergic,
cortical neurons. Activation of CRHR1 in the forebrain is associated with
alteration in glutamatergic neurotransmission and increased behavioral
susceptibility to stress in mice. Therefore, our single-cell findings support
our hypothesis associating cortical CRHR1 expression with increased stress
susceptibility, cortical thinning, and alcohol misuse."
https://jamanetwork.com/journals/jamapsychiatry/articlepdf/2795312/jamapsychiatry_mavromatis_2022_oi_220047_1662054847.3687.pdf
[2140]
Durazzo et al (2023) concentrated on cortical
thickness in alcohol withdrawal, but for a longer period than previous attempts.
As the authors explained:
"Cortical thickness may show a differential pattern of
recovery with abstinence in alcohol use disorder (AUD) compared to volume and
surface area measures in the same brain regions…The cerebral cortex is primarily
composed of neuronal and glial cells [i.e., astrocytes, oligodendrocytes, and
microglia…and the ratio of glial cells to neurons is approximately 0.7:1;
accordingly, cortical thickness may serve as a macroscopic surrogate marker of
the cytoarchitectural integrity of cells comprising the cortex."
So
"AUD participants were studied at approximately 1 week
(n=68), 1 month (n=88) and 7.3 months (n=40) of abstinence.
"Forty-five never-smoking controls (CON) completed a
baseline study, and 15 were reassessed after approximately 9.6 months.
Participants completed magnetic resonance imaging studies at 1.5T and cortical
thickness for 34 bilateral regions of interest (ROI) was quantitated with
FreeSurfer. AUD demonstrated significant linear thickness increases in 25/34 ROI
over 7.3 months of abstinence."
But the
"...rate of change from 1 week to 1 month was greater
than 1 month to 7.3 months in 19/34 ROIs."
Overall
"After 7.3 months of abstinence, AUD were
statistically equivalent to CON on cortical thickness in 24/34 ROIs; the
cortical thickness differences between AUD and CON in the banks superior
temporal gyrus, post central, posterior cingulate, superior parietal,
supramarginal and superior frontal cortices were driven by thinner cortices in
AUD with proatherogenic conditions relative to CON. In actively smoking AUD,
increasing pack-years was associated with decreasing thickness recovery
primarily in the anterior frontal ROIs..."
and
"Widespread bilateral linear cortical thickness
recovery over 7.3 months of abstinence was the central finding for this AUD
cohort. Proatherogenic conditions were associated with decreased thickness
recovery and thinner cortex after 7.3 months of abstinence in several ROIs; this
suggests alterations in perfusion or vascular integrity may relate to structural
recovery in AUD. These results support the adaptive and beneficial effects of
sustained sobriety on brain structural recovery in those with AUD."
https://www.sciencedirect.com/science/article/abs/pii/S074183292300263X
[4191]
Shapson-Coe et al (2024) found a different glia to
neuron ratio in their 1 cubic mm sample:
"To fully understand how the human brain works,
knowledge of its structure at high resolution is needed. Presented here is a
computationally intensive reconstruction of the ultrastructure of a cubic
millimeter of human temporal cortex that was surgically removed to gain access
to an underlying epileptic focus. It contains about 57,000 cells, about 230
millimeters of blood vessels, and about 150 million synapses and comprises 1.4
petabytes. Our analysis showed that glia outnumber neurons 2:1, oligodendrocytes
were the most common cell, deep layer excitatory neurons could be classified on
the basis of dendritic orientation, and among thousands of weak connections to
each neuron, there exist rare powerful axonal inputs of up to 50 synapses.
Further studies using this resource may bring valuable insights into the
mysteries of the human brain."
https://www.science.org/doi/10.1126/science.adk4858 [4641]
Alcohol is not a drug, it's a drink. So we don't need
to worry about these brain volumes or cortical thicknesses in Ptuj, where I
think you would struggle to find 40 people who didn't have a drink for 7.3
months.
It is sufficient to note that any change in cortical
thickness from cannabis alone is a separate and distinct effect from cortical
thinning caused by alcohol consumption.
Owens et al (2022) say
"Cortical thinning in adolescence is well-established
as a normal trajectory of brain development. Studies estimate cortical thinning
of around 1% annually, which comes out to around 0.03–0.06 millimeters per
year."
Moreover there is research
"...suggesting that the accelerated thinning may be
mediated, in part, by cannabis exposure affecting the brain’s endogenous
cannabinoid system. That said, it remains possible that these brain changes may
not be a consequence of the cannabis exposure but may reflect instead a
neurodevelopmental trajectory caused by other factors that is related to a
higher likelihood of adolescent cannabis use."
https://www.nature.com/articles/s41398-022-01956-4 [4193]
The good news is that marijuana use appears, for
whatever reason, to ameliorate cortical thinning in drinking teenagers. From the
perspective of this metric you are better off with both than alcohol alone.
In "Adolescent Cortical Thickness Pre- and Post
Marijuana and Alcohol Initiation" by Jacobus et al (2016):
"Adolescents (N=69) were followed from ages 13
(pre-initiation of substance use, baseline) to ages 19 (post-initiation,
follow-up). Three subgroups were identified, participants that initiated alcohol
use (ALC, n=23, >20 alcohol use episodes), those that initiated both alcohol and
marijuana use (ALC+MJ, n=23, >50 marijuana use episodes) and individuals that
did not initiate either substance regularly by follow-up (CON, n=23, <3 alcohol
use episodes, no marijuana use episodes). All adolescents underwent
neurocognitive testing, neuroimaging, and substance use and mental health
interviews."
This is their Figure 2:
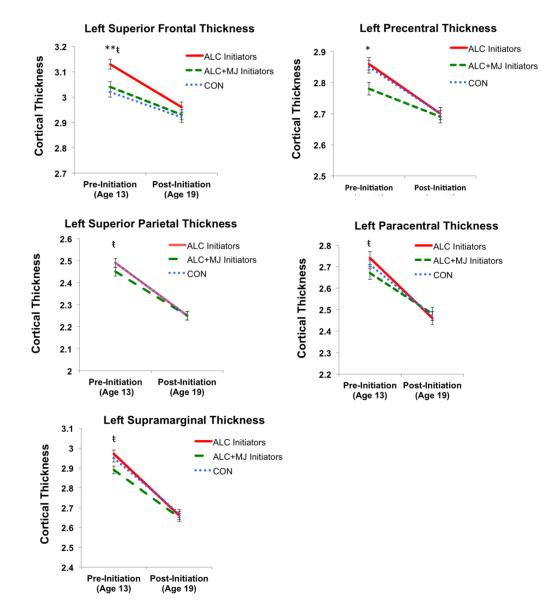
"Examination of 34 independent cortical regions in
each hemisphere revealed significant Group by Time effects, largely consistent
with a significant decrease in cortical thickness over time in the ALC group
compared to the ALC+MJ group (ps<.05). Findings within each lobe of the brain
are presented below (see Figures 2--4).4). Intracranial volume (ICV) was
identified a priori as a covariate."
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5072451/ [4192]
If you are banning marijuana for reasons of cortical
thickness, it only raises questions as to why there is no minimum age for
drinking alcohol in public in Slovenia, since these authors didn't even bother
to find a marijuana-only cohort, suggesting alcohol might be a gateway drug. I
think we can also conclude these studies were not as advanced in 1925, 1961,
1971 and 2000, as they today are, and this topic played no role in the
prohibition of cannabis.
Despite this, unlike our alcoholic cousins, cannabis
users know very well that THC isn't for everyone. Who knew? Where is the
equivalent analysis for wine? Can we find more than associations. Can we
actually say there are genes which CAUSE people to drink?
With a lot of data and math, we can. After finding
correlations between schizophrenia and opioids, nicotine, cannabis and - most of
all - alcohol...
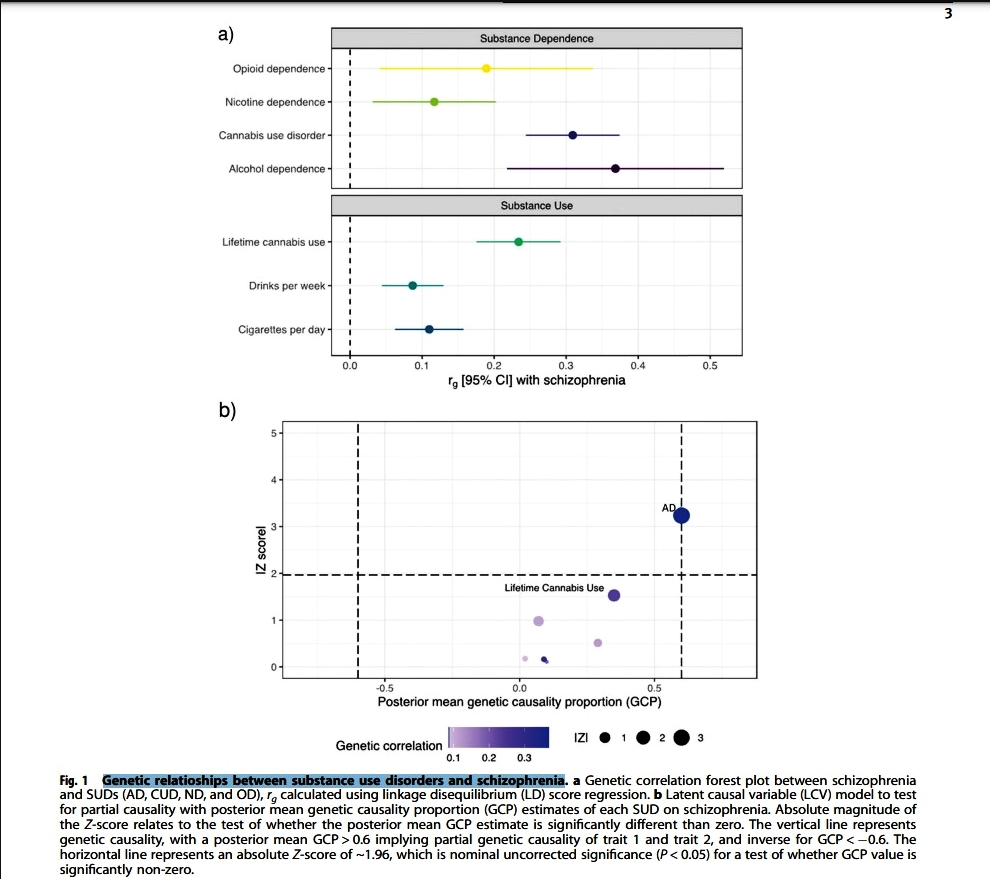
...Greco et al in Translational Psychiatry (2022)
write:
"Evidence for partial genetic causality of alcohol
dependence on schizophrenia LD score regression (LDSR) analysis revealed
significant genetic correlations between schizophrenia and several of the SUD
phenotypes after correcting for the number of tests performed (Fig. 1a).
Specifically, schizophrenia was positively genetically correlated with AD (rg =
0.368, SE = 0.076, P = 1.61 × 10−6 ), CUD (rg = 0.309, SE = 0.033, P = 1.97 ×
10−20), ND (rg = 0.117, SE = 0.043, P = 7.0 × 10−3 ), and the substance use
phenotypes CPD (rg = 0.11, SE = 0.024, P = 4.93 × 10−6 ), DPW (rg = 0.087, SE =
0.021, P = 6.36 × 10−5 ) and LCU (rg = 0.234, SE = 0.029, P = 3.74 × 10−15). We
also found nominally positive genetic correlation between schizophrenia and OD
(rg = 0.184, SE = 0.075, P = 0.0142), however, this did not survive
multiple-testing correction. LCV models were then constructed for the
significant traits for which their schizophrenia genetic correlation estimate
passed multiple-testing correction (AD, CUD, ND, CPD, DPW, and LCU) to
investigate whether any of the observed genetic correlations between SUD and
schizophrenia may constitute a causal relationship (Fig. 1b, Supplementary Table
1). There was no evidence for partial genetic causality of CUD, ND, CPD, DPW,
and LCU on schizophrenia, but there was moderate evidence that AD was partially
genetically causal for schizophrenia (GCP = 0.60, SE = 0.22, P = 0.001). We note
that while the SNP heritability estimate for AD was significantly non-zero, the
Z-score for AD (h2 / SE) was somewhat noisier (Zh 2 = 5.98) than recommended by
the authors of the LCV method (Zh 2 > 7). As a result, this inference of partial
genetic causality needs to be cautiously interpreted in light of this, with
larger GWAS of AD diagnosed using DSM-IV/DSM-V or similar likely required to
boost the precision of AD SNP heritability. DPW did not show any evidence for a
causal relationship with schizophrenia like alcohol dependence. Interestingly,
AD and DPW showed genetic correlation (rg = 0.709, SE = 0.105, P = 1.38 ×
10−11), as did CUD and LCU (rg = 0.476, SE = 0.049, P = 2.71 × 10−22) but there
was no evidence of partial genetic causality of DPW on AD (GCP = 0.05, SE =
0.56, P = 0.88) or CUD and LCU (GCP = 0.02, SE = 0202, P = 0.976). No evidence
of genetic correlation was observed among ND and CPD (rg = 0.071, SE = 0.054, P
= 0.192). These results suggest that the underlying mechanisms driving AD, CUD,
and ND may not strongly present in substance use observed in a population
sample, although this requires further investigation."
With this collection of correlations Greco et al go on
to reveal some evidence for causation:
"In the pairwise meta-analysis, 73 gene-sets were
statistically significant (FDR < 0.05), for schizophrenia meta-analysed with AD,
with 10 gene-sets not previously observed in the individual schizophrenia GWAS
(Supplementary Table 10), including longterm synaptic potentiation (ngenes = 81,
P = 5.71 × 10−5, FDR = 0.02), exocytic vesicle (ngenes = 196, P = 1.23 × 10−4,
FDR = 0.03), paroxysmal ventricular tachycardia (ngenes = 23, P = 1.70 × 10−4,
FDR = 0.03), peptidyl serine dephosphorylation (ngenes = 19, P = 1.75 × 10−4,
FDR = 0.04), hypoplasia of the olfactory bulb (ngenes = 4, P = 2.12 × 10−04, FDR
= 0.04), regulation of heart contraction (ngenes = 221, P = 2.32 × 10−4, FDR =
0.04), and regulation of peptidyl serine dephosphorylation (ngenes = 5, P = 2.32
× 10−4, FDR = 0.04)."
"We further constructed latent causal variable (LCV)
models to test for partial genetic causality and found evidence for a potential
causal relationship between alcohol dependence and schizophrenia (GCP = 0.6, SE
= 0.22, P = 1.6 × 10−3 ). This putative causal effect with schizophrenia was not
seen using a continuous phenotype of drinks consumed per week, suggesting that
distinct molecular mechanisms underlying dependence are involved in the
relationship between alcohol and schizophrenia. To localise the specific genetic
overlap between schizophrenia and substance use disorders (SUDs), we conducted a
gene-based and gene-set pairwise meta-analysis between schizophrenia and each of
the four individual substance dependence phenotypes in up to 790,806
individuals. These bivariate meta-analyses identified 44 associations not
observed in the individual GWAS, including five shared genes that play a key
role in early central nervous system development. The results from this study
further supports the existence of underlying shared biology that drives the
overlap in substance dependence in schizophrenia, including specific biological
systems related to metabolism and neuronal function."
and
"We then considered the association of 2,598 microRNA
(miRNA) regulatory target prediction gene-sets with each individual GWAS,
followed by the bivariate meta-analyses (Supplementary Tables 14–18). There were
239 miRNA that passed FDR correction for the individual schizophrenia GWAS, no
miRNA regulator target genesets passed FDR correction for any of the individual
substance dependence phenotypes. Notably, each of the meta-analyses revealed a
total of 17 microRNA regulatory target gene-sets, not seen in the individual
phenotypes, including six found in more than one bivariate meta-analysis. One
such interesting example was the predicted target genes of miR-495, a microRNA
that is highly enriched in the nucleus accumbens and has been shown to play a
role in addiction-related behaviours. MiR-495 survived correction in both
schizophrenia and AD meta-analysis (ngenes = 231, P = 2.43 × 10−3, FDR = 0.03),
and the schizophrenia and ND model (ngenes = 231, P = 5.88 × 10−5, FDR = 0.01)
but was only nominally significant in the individual schizophrenia GWAS (P =
0.007), supporting how this meta-analysis approach can increase discovery
power."
The authors discuss this
"...further evidence of a causal relationship between
alcohol dependence and schizophrenia. Interestingly, there was no causal
relationship between the consumption of alcohol (drinks per week) and
schizophrenia, or between AD and DPW [drinks per week]. This was consistent with
the transancestral GWAS [genome-wide association study] of alcohol dependence,
which suggested there is a distinction in the underlying molecular mechanisms
driving pathological and non-pathological behaviours for substance use and
dependence, particularly within biological pathways implicated in the
psychopathological aspects of problematic drinking. Additionally, it is also
well known that psychotic symptoms can occur in several clinical conditions
related to alcohol such as intoxication, withdrawal, alcohol-induced psychotic
disorder, and delirium. Although registry data-sets come with several
limitations such as the threat of false-negatives due to under-reporting of
substance use, a study on 18,478 Finnish inpatients found alcohol-induced
psychosis was the most common type of substance-induced psychotic disorder
(SIPD), with a separate Swedish study that followed 7606 individuals for 84
months between 1995 and 2015 found that for alcohol the risk for SIPD was 4.7%.
Interestingly, 22.1% (95% CI = 17.6−27.5) of patients who had previously
received a diagnosis of alcohol-induced psychosis went on to develop
schizophrenia. The putative causal relationship of AD on schizophrenia warrants
further epidemiological and biological interrogation. There are also some
important limitations to the use of LCV [latent causal variable]
models—specifically, they are bivariate in nature, and thus, cannot model the
effect of other plausible mediators or confounders, while the posterior mean GCP
[gradient conjugate prior] estimate is also not a causal estimate that could be
afforded by approaches like Mendelian randomisation. However, the use of
Mendelian randomisation with a binary exposure like AD can be challenging,
particularly as only a handful of genome-wide significant SNPs have been
identified that could be suitable instrumental variables."
https://www.nature.com/articles/s41398-022-02186-4.pdf [1901]
The key idea of Mendelian randomisation is that, per
Mendel's Law of Independent Assortment, the random assignment of genetic
variants during meiosis mimics the randomisation that occurs in a randomised
control trial. All confounders (biological and social factors) are otherwise
balanced if we study people according to their allocated genetic variants,
"The use of the terms loci, alleles, genes, genotype,
and polymorphisms has evolved since Mendel’s use of ‘differentiating
characteristics’, and conventions in usage differ between human and animal
geneticists, which adds to confusion. For clarity, these terms are defined (Box
4, Figure 2) [below]. Briefly, the genotype of an individual refers to the two
alleles inherited at a specific locus—if the alleles are the same, the genotype
is homozygous, if different, heterozygous. A polymorphism is the existence of
two or more variants (e.g. SNPs) at a locus. The basic ideais that, if such
polymorphisms produce phenotypic differences that mirror the biological effects
of modifiable environmental exposures which in turn alter disease risk, the
different polymorphisms should themselves be related to disease risk to the
extent predicted by their influence on the phenotype. Common polymorphisms that
have a well-characterized biological function can therefore be utilized to study
the effect of a suspected exposure on disease risk. One key point is that the
distribution of such polymorphisms is largely unrelated to the sorts of
confounders—socioeconomic or behavioural—that were identified above as having
distorted interpretations of findings from observational epidemiological
studies."
And their glossary of genetic terms:
"alleles are the variant forms detectable at a locus
canalization is the process by which potentially
disruptive influences on normal development from genetic (and environmental)
variations are damped or buffered by compensatory developmental processes
a gene comprises a DNA sequence, including introns,
exons, and regulatory regions, related to transcription of a given RNA
genotype of an individual refers to the two alleles
inherited at a specific locus—if the alleles are the same, the genotype is
homozygous, if different, heterozygous
a haplotype is the set of alleles present at a series
of linked loci on a chromosome; a person has two haplotypes for any such series
of loci, one inherited maternally and the other paternally
linkage disequilibrium is the association between
alleles at different loci within the population. Linkage disequilibrium can
exist because alleles are physically close together and tend to be co-inherited,
or because they occur together for reasons of population origin in subsections
of an overall population and therefore demonstrate a statistical association
within the overall population
a locus is the position in a DNA sequence and may be
used to refer to a single nucleotide polymorphism (SNP), or to larger regions of
DNA sequence
a marker is a segment of DNA with an identifiable
physical location on a chromosome, whose inheritance can be followed and can be
assayed in genetic association studies. In such studies, markers are of interest
if they are linked to polymorphisms with functional significance. A marker can
be a gene, an SNP or a section of DNA with no known function
a mutation is a permanent structural alteration in DNA
or the process by which a DNA sequence is altered
pleiotropy is the potential for polymorphisms to have
more than one specific phenotypic effect
polymorphism is the existence of two or more variants
at a locus. Conventionally, the prevalence in the population should be above 1%
to be referred to as a polymorphism; if prevalence is below this, variants are
referred to as mutations
population stratification is an example of confounding
in which the co-existence of different disease rates and allele frequencies
within population sub-sections lead to an association between the two at a whole
population level
single nucleotide polymorphisms (SNPs) are positions
along a chromosome where the genetic code varies between individuals by a single
base pair (pronounced ‘snips’)"
https://academic.oup.com/ije/article/32/1/1/642797?login=false [1960]
The verbal idea of characteristics "Mendelizing" can
be traced back to Morgan in 1913.
https://archive.org/details/hereditysex00morg/page/87/mode/1up?ref=ol&view=theater&q=Mendelize
[1960]
Havard epidemiologists O'Connor and Price describe the
LCV model:
"The latent causal variable (LCV) model is based on a
latent variable L that mediates the genetic correlation between the two traits
(Figure 1a). Under the LCV model, trait 1 is fully genetically causal for trait
2 if it is perfectly genetically correlated with L; ``fully" means that the
entire genetic component of trait 1 is causal for trait 2 (Figure 1b). More
generally, trait 1 is partially genetically causal for trait 2 if the latent
variable has a stronger genetic correlation with trait 1 than with trait 2;
``partially" means that part of the genetic component of trait 1 is causal for
trait 2. In order to quantify partial causality, we define the genetic causality
proportion (gcp) of trait 1 on trait 2. The gcp ranges between 0 (no partial
genetic causality) and 1 (full genetic causality). A high value of gcp (even if
it is not exactly 1) implies that interventions targeting trait 1 are likely to
affect trait 2. An intermediate value implies that some interventions targeting
trait 1 may affect trait 2....We caution that low gcp estimates are not evidence
of full genetic causality, and we refer to trait pairs with low gcp estimates as
having limited partial genetic causality. LCV p-values test the null hypothesis
that gcp=0, and a highly significant p-value does not imply a high gcp."
https://europepmc.org/backend/ptpmcrender.fcgi?accid=PMC6684375&blobtype=pdf
[1962]
According to Lu Qi of the Department of Nutrition,
Harvard School of Public Health, Boston and elsewhere:
"The Mendelian randomization approach also holds
considerable promise in the study of intrauterine influences on offspring health
outcomes."
and
"Mendelian randomization studies may have particular
relevance in assessing the effects of long-term (lifetime) exposures, such as
dietary intake, lifestyle, and excessive adiposity, whereas RCTs can examine
only short-term effects. Interest in using Mendelian randomization in causality
inference has been growing rapidly over the past 5 years."
and has a little more on the maths:
"Recently, the instrumental variable (IV) method was
introduced as a formal test of Mendelian randomization using the genetic markers
as IVs. This method calculates the predicted effect of exposure on outcome,
given the associations between genotype and exposure and between genotype and
outcome. The IV approach is widely used in econometrics to deal with
'endogeneity,' a broad term covering confounding, reverse causality, and
regression dilution bias. An IV should be: 1) not correlated with the
confounders; 2) related to the exposure of interest; and 3) related to the
outcome only through the exposure of interest. In the context of Mendelian
randomization analysis, the genetic variant, which is not associated with
confounding variables, can act as an IV for the environment exposure. The
two-stage least squares (2SLS) method is most widely used to fit the IV models.
In brief, in the first stage of the analysis, IV is used to generate predicted
values for the independent exposure (endogenous) variable. The predicted value
is then used in a second-stage regression to explain the variation in outcome.
The F-statistics from the first-stage regressions can be used to evaluate the
strength of the IV in estimating the exposure. A value greater than 10 is
generally assumed to be a sign of sufficient strength. Because genotype is
associated with the exposure but not the confounders, the predicted
exposure–outcome association is not confounded and is likely the estimate for
the causal effect. Other methods such as a structural equation model have also
been used in Mendelian randomization analysis. In the case of non-linear
variables, the structural equation approach allows a simpler interpretation of
the regression coefficients when compared with an IV approach."
Mendelian randomization has supported causality in
some associations, and abolished it in others (Table 1). They describe how
genetic risk factors for esophegal cancer were calculated:
"Alcohol consumption has been related to the risk of
esophageal cancer, partly through the carcinogenic effects of alcohol’s
principal metabolite acetaldehyde. However, alcohol intake tends to be
correlated with other lifestyle risk factors such as smoking and diet. Lewis et
al. examined the causal relation between alcohol and esophageal cancer risk
using genetic data on the ALDH2 (aldehyde dehydrogenase 2 family) gene, which
encodes the major enzyme eliminating acetaldehyde. A single point mutation in
ALDH2 results in the 2*2 allele (glutamic acid to lysine substitution at residue
487) and inactivation of the enzyme. This in turn leads to an accumulation of
acetaldehyde after alcohol intake. Individuals who are homozygous for the 2*2
allele have peak blood alcohol levels that are 18 times higher and heterozygotes
5 times higher than *1*1 homozygotes. Drinking alcohol is likely to cause
unpleasant symptoms such as nausea, drowsiness, and headache in the carriers.
Therefore, *2*2 homozygotes drink significantly less alcohol than the wild-type
carriers, and heterozygotes fall somewhere in the middle. The authors performed
a meta-analysis including 905 cases of esophageal cancer and 2,078 controls from
seven studies carried out in Japan, Taiwan, and Thailand. The relative risk (RR)
of esophageal cancer for *1*1 homozygotes versus *2*2 homozygotes was 2.77 [95%
confidence interval (CI), 1.25–6.12]. The authors predicted RR for genotype *1*1
based on the following data: 1) A meta-analysis that found light, moderate, and
heavy drinkers had 1.8-, 2.38-, and 4.36-fold higher risk of cancer,
respectively, than nondrinkers.27 2) One study reported that 9.4%, 28.2%, 39.6%,
and 22.9% of *1*1 individuals were non-, light, moderate, and heavy drinkers,
respectively; whereas all 2*2 individuals were virtually nondrinkers. The
overall RR for *1*1 homozygotes was then derived with a formula RR = RRi × Pi,
where i denotes the drinking category (non-, light, moderate, or heavy), RRi is
the relative risk in the ith drinking category estimated by a meta-analysis, and
Pi is the assumed proportion of ith drinking category among controls. The
predicted RR was 2.54, comparable to the meta-analysis on the non-confounded
genetic association (RR = 2.77). The data support a causal effect of drinking on
esophageal cancer risk."
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3671930/ [1961]
Continuing with the drinking:
In Kapoor et al: "Multi-omics integration analysis
identifies novel genes for alcoholism with potential overlap with
neurodegenerative diseases" in Nature Communications 2021, the authors looked
for causal variants and genes...underlying the biology of alcohol use disorder
(AUD) and drinks per week (DPW):
"Using Mendelian Randomization-based methods on the
largest available transcriptomic and epigenomic data for brain tissues
(Supplementary Data 1) and myeloid cells, we prioritized regulatory variants
that influence AUD and DPW (Fig. 1). The current manuscript explored the
multi-omic integration results in AUD and DPW separately and subsequently
focused on the overlapping genes between these two traits. Overlapping genes
prioritized in current analysis are primarily driven by individual GWASs.
Therefore, these signals are minimally influenced by the sample size bias that
might arise due to the integration of two correlated traits with extreme
differences in power....To the best of our knowledge, this is the largest
systematic multi-omics integration analysis to identify the functional impact of
variants and genes associated with two correlated but etiologically distinct
aspects of alcohol involvement."
Among the results:
"AUD meta-analysis. The large meta-analysis of AUD
GWAS summary statistics (N = 48,545 AUD cases and 187,065 controls) from the
Million Veterans Program (MVP)19, the Psychiatric Genetics consortium
(PGC-SUD)12 and the Collaborative Studies on Genetics of Alcoholism (COGA)20
identified 1157 SNPs (31 independent lead SNPs) within or near 79 genes at 10
independent loci associated with AUD (Supplementary Figs. 1–4). We did not
include UKB-AUDIT-P in this meta-analysis to specifically focus on AUD. Many of
these loci were shared between the AUD GWAS meta-analysis and the DPW GWAS by
Liu et al. who identified 81 independent loci represented by 5197 (>200
independent lead) SNPs. A total of 360 SNPs associated with AUD and DPW were in
common (i.e., p < 5 × 10−8 in both GWAS) (Supplementary Fig. 5). A large and
nominally significant proportion (45%) of AUD and DPW-associated SNPs were
within intronic, UTR and non-coding regions of the genome (Supplementary Fig.
6). LDSC analysis using tissue specific epigenetic annotations. We used the
stratified linkage disequilibrium score (LDSC) regression to test whether the
heritability of AUD and DPW is enriched in regulatory regions surrounding genes
in a specific tissue. Using multi-tissue chromatin (ROADMAP and ENTEX) data, we
observed a significant enrichment of promoter-specific epigenetic markers
(H3K4me1/me3) in the fetal and the adult (germinal matrix, frontal-cortex) brain
(P < 5 × 10−8) (Fig. 2; Supplementary Data 2 and 3) for the SNPs associated with
AUD and DPW, respectively."
and
"Identification of SPI1 and MAPT as genes for AUD are
good examples of pleiotropy and/ or causal links between the alcohol intake and
susceptibility to AUD, other psychiatric disorders (e.g., depression), and even
Alzheimer’s disease and other neurodegenerative diseases. We found that
increased SPI1 expression in myeloid lineage cells was associated with a higher
DPW and higher risk for AUD. Recently, Zhang and colleagues observed that
protein expression levels of SPI1 in the cerebellum and spleen from subjects
with Major depressive disorder and schizophrenia were significantly higher than
in controls. In the past, we have demonstrated that functional variants related
to SPI1 expression are associated with the risk of Alzheimer’s disease. Similar
to this study, higher levels of expression of SPI1 is associated with increased
risk for Alzheimer’s disease. SPI1 (Spi-1 Proto-Oncogene) encodes an ETS-domain
transcription factor (PU.1) that regulates gene expression during myeloid and
B-lymphoid cell development and homeostasis. This nuclear protein binds to a
purine-rich sequence known as the PUbox found near the promoters of target genes
and, in coordination with other transcription factors and cofactors, regulates
their expression; among the genes are LXR/RXR nuclear receptors. In the brain,
SPI1 is specifically expressed in microglia. Given SPI1’s control over
expression of several downstream genes, this gene may be a major reason
enrichment of immune pathways is observed in transcriptomic analysis of human
and animal brains. Because of the small fraction of microglia in bulk brain
tissue, it is difficult to study the expression of this transcription factor in
transcriptomic datasets from whole brains. Some studies using animal models have
reported that chronic alcohol consumption can influence the expression of PU.1
in isolated microglia and peripheral lung macrophages. However, these studies
report the consequences of drinking on PU.1 expression whereas our study uses
genomic evidence to demonstrate that regulation of innate immune response likely
underlies, at least in part, susceptibility to increased drinking and eventual
risk for AUD. MAPT is another example of a pleiotropic relationship between AUD
and other neuropsychiatric and neurodegenerative disorders. Located on
chromosome 17, MAPT, encodes the tau proteins best known medically for their
role in central nervous system disorders such as Alzheimer’s disease,
frontotemporal dementia, Parkinson’s disease, and the primary tauopathies
progressive supranuclear palsy and corticobasal degeneration. Recently, Hoffman
and colleagues showed that alcohol use can upregulate the expression of pTau
(Ser199/Ser202) in the hippocampus of C57BL/6J mice. Another study in humans
observed differences in CSF-Tau levels in demented people with alcohol use vs
Alzheimer disease patients. CRHR1 (corticotropinreleasing hormone type I
receptor) is another gene on 17q.21.31, that has been reported to be associated
with alcoholism. However, in our analysis, we did not observe an association
between CRHR1 expression and alcohol consumption. We also identified other genes
that might be involved in increased alcohol consumption through a variety of
biological mechanisms. For example, VPS4A at 16q23.1 has been implicated in
dopamine regulation, reward anticipation, and hyperactivity in an fMRI study. We
also identified functional variants for SULT1A1 and SULT1A2 genes that encode
for Sulfotransferase Family 1A enzymes catalyzing the sulfate conjugation of
many hormones, neurotransmitters, drugs, and xenobiotic compounds. In IPA
disease enrichment analysis, we observed a nominally significant overlap between
genes implicated in DPW with other neurological, behavioral and immune-related
disorders (Supplementary Fig 16). The genes associated with DPW also showed
significant enrichment for pathways related to TR/ RXR activation, Lipoate
biosynthesis, Estrogen biosynthesis, and Sirtuin signaling (Supplementary Data
8). TRs (Thyroid hormone receptor) control the expression of target genes
involved in diverse physiological processes and diseases, such as metabolic
syndrome, obesity, and cancer, and, therefore, are considered as important
targets for therapeutic drug development. RXRs (Retinoic X Receptor) are known
to potentially regulate the ethanol metabolizing enzymes after chronic alcohol
consumption. It has been reported that the human aldehyde dehydrogenase-2
(ALDH2) promoter contains a retinoid response element, which might be
contributing to the regulation of the gene. Sirtuins signaling has been shown to
play an important role in cocaine and morphine Action in the Nucleus Accumbens.
Ferguson and colleagues demonstrated that systemic administration of a
nonselective pharmacological activator of all sirtuins can increase the cocaine
reward."
"We have identified a number of candidate causal genes
for DPW and AUD, resulting from a multi-omic analysis of human genetic and
expression data."
Again attention is drawn to the lack of any practical
use of this data in treating the individual with the information available to
him or her under the present circumstances. What the everyday drinker or his bar
knows about, say, Slovenia's predisposing genes, is negligible to nothing.
https://www.researchgate.net/journal/Nature-Communications-2041-1723/publication/354030227_Multi-omics_integration_analysis_identifies_novel_genes_for_alcoholism_with_potential_overlap_with_neurodegenerative_diseases/links/61208e290c2bfa282a5ce4ca/Multi-omics-integration-analysis-identifies-novel-genes-for-alcoholism-with-potential-overlap-with-neurodegenerative-diseases.pdf
[1902]
And let's not forget "Transancestral GWAS of alcohol
dependence reveals common genetic underpinnings with psychiatric disorders"
(2018) in which Walters et al remind us that
"Liability to alcohol dependence (AD) is heritable,
but little is known about its complex polygenic architecture or its genetic
relationship with other disorders. "
"The current analysis identified robust genetic
correlation of AD with a broad variety of psychiatric outcomes. This correlation
was strongest for aspects of negative mood, including neuroticism and major
depression, as also seen in twin studies and through recent specific molecular
evidence for pleiotropy. Taken together with evidence from other recent genomic
studies, and with null correlations for other GWAS of alcohol consumption but
not for measures of problem drinking (for example, AUDIT-P), these findings
suggest that major depression may primarily share genetic liability with alcohol
use at pathological levels.
"AD was also strongly genetically correlated with poor
educational and socioeconomic outcomes and marginally correlated with measures
of risk-taking. Nominally significant genetic correlations with delay
discounting (i.e., favoring immediate rewards) and risktaking, and the strong
genetic correlation of AD with attention deficit–hyperactivity disorder,
cigarette smoking, and cannabis use, may similarly reflect a shared genetic
factor for risk-taking and reduced impulse control. Common genetic liability to
early, risky behaviors is characteristic of both AD and age of first birth. The
observed negative genetic correlation with age of first birth is consistent both
with risk-taking and with the significant genetic correlations of AD with lower
socioeconomic status, as indexed by higher neighborhood Townsend deprivation
score and lower educational attainment. Lower socioeconomic status is correlated
with both AD and age of first birth, and the current study suggests that shared
genetic liabilities may be one potential mechanism for their observed
relationship. However, the question of whether these genetic correlations
represent causal processes, horizontal pleiotropy, or the impact of unmeasured
confounders should be explored in the future.
"Lower genetic correlations were observed for most
biomedical and anthropometric outcomes. Liver enzymes GGT and ALT, once proposed
as possible biomarkers for alcohol abuse, showed only nominal evidence for
genetic correlation with AD, and neither survived multiple-testing correction.
Notably, we did not find any association between AD and body-mass index (BMI).
Negative genetic correlations with BMI were previously reported for both alcohol
consumption and AUDIT scores, but there is prior evidence that BMI has differing
underlying genetic architectures in the context of AD and outside of that
context. The negative genetic correlations observed in those studies are
consistent with studies of light to moderate drinking, which is also associated
with healthier lifestyle behaviors, while heavy and problematic drinking is
typically associated with weight gain."
https://helda.helsinki.fi/bitstream/10138/307699/1/s41593_018_0275_1.pdf
[1904]
Philip C Haycock thinks the concept of a “fetal
alcohol spectrum” should be expanded to include “preconceptional effects.”
Writing in 2009, he references not the latest research, but some animal
experiments published in 1913 which are very germane to this preconceptual
effect theory. The fetus is not directly exposed to the toxin at all,
Dr Stockard's paper "The Effect on the Offspring of
Intoxicating the Male Parent and the Transmission of the Defects to Subsequent
Generations" is still widely cited today:
"In one extensive series of experiments, guinea pigs
were treated by the inhalation method to the point of intoxication every day,
except Sundays, for approximately 3 yr. 'From time to time,' treated animals
(males and females) were mated with untreated control. Various experimental
conditions were tested, such as 'alcoholized females' × 'normal males'
'alcoholized females' × 'alcoholized males,' and 'alcoholized males' × 'normal
females.'
"It was found that after 34 successful crossings
between alcoholized males and normal females, 24% of litters were stillborn. The
remaining litters produced 54 offspring, 39% of which died soon after birth. In
comparison, a normal male × normal female crossing resulted in 33 litters, of
which 1 (3%) was stillborn, and of the 60 live offspring, 4 (7%) died soon after
birth. In addition, the untreated offspring of parents from alcoholized
conditions tended to have fewer surviving offspring than controls (54% vs. 93%).
In sum, these results suggest that alcohol administered to males during the
preconceptional period resulted in high rates of perinatal mortality in
offspring, and that these effects persisted into the F(2) generation."
https://www.journals.uchicago.edu/doi/epdf/10.1086/279379 [1905]
After summarising various unpleasant physiological
preconceptual effects, upon the offspring, of alcohol administered to daddy rats
prior to conception, Haycock turns to behaviour.
"Studies employing chronic dosage regimens have also
uncovered behavioral effects of paternal alcohol exposure. For example, in one
study, 18-day-old offspring of male rats intubated with 3 or 2 g/kg ethanol
twice a day for 7 mo were more active in the open field compared with controls,
and they took a greater number of trials to complete a passive avoidance
learning task.
"In another study, male mice were maintained on a
liquid diet for 56–61 days in which alcohol provided 0%, 10%, or 20% of their
calories. Organ weights (except for thymus), litter size, and weights at birth
and at ages 21 and 55 days were not affected. However, a dose-dependent decrease
in physical activity at 20 and 24 days of age, as well as decreases in serum
testosterone at 55 days of age, were observed in offspring derived from the
alcohol treatment groups. In addition, offspring of fathers receiving 20% of
their calories from alcohol performed better on a passive avoidance task but
more poorly in a T-maze task.
"In a study of both mice and rats, males received 0%,
10%, or 25% of the calories from ethanol during 7–14 wk. It was observed that
mice from the alcohol-exposed group were relatively more immobile in a swimming
behavioral task. In contrast, rats from the alcohol-exposed group displayed an
opposite effect, being less immobile in the same task.
"In a separate study, male rats receiving 17.5% or 35%
of their dietary calories from alcohol sired female offspring who performed
worse in a two-way shock avoidance learning task. However, no effects on birth
weight, spontaneous alternation, or passive avoidance learning were observed.
"Studies employing acute dosage regimens have also
reported effects of paternal alcohol exposure on offspring development. In one
study, in which male rats were intubated with a once-off dose of either 6, 4, 2,
or 0 g/kg ethanol, there was a significant dose-response effect on the frequency
of runts (<5.5 g at birth) and the number of malformations. However, no
differences in mating, fecundity, or litter size were reported. In a separate
study, a once-off dose of 5 g/kg administered by intraperitoneal injection to
male rats 24 h prior to mating resulted in fewer matings, smaller litter sizes,
and higher fetal mortality."
Much more in the article about the possible
mechanisms. He concludes:
"In sum, alterations in epigenetic programming may
underlie the teratogenic consequences of ethanol exposure prior to conception,
as well as after conception, during preimplantation and gastrulation. One of the
main implications of an epigenetic perspective is that the FASD spectrum is not
limited to clinical defects arising from in utero ethanol exposure, suggesting
that the concept of a fetal alcohol spectrum should be expanded to include
preconceptional effects."
and
"Finally, an epigenetic perspective suggests that
alcohol exposure outside of the organogenic period (e.g., during preimplantation
or prior to conception) might have teratogenic consequences for the CNS. Indeed,
the association of paternal alcohol consumption with behavioral and cognitive
abnormalities in offspring in some animal and human studies (discussed above)
supports this view. Because such cases are unlikely to receive a diagnosis
within the FASD spectrum (gestational alcohol exposure being a requirement for
diagnosis), this raises the possibility that transgenerational responses to
alcohol might account for a significant proportion of idiopathic
neurodevelopmental disorders (e.g., idiopathic autism) in humans."
https://academic.oup.com/biolreprod/article/81/4/607/2557677 [1906]
Burdinski et al (2024) longitudinally measured medical
cannabis users' task abilities and brain volumes using fMRI.
"No statistically significant difference in brain
activation between the 2 time points (baseline and 1 year) in those with medical
cannabis cards and no associations between changes in cannabis use frequency and
brain activation after 1 year were found."
https://jamanetwork.com/journals/jamanetworkopen/fullarticle/2823671 [3550]
In the years since the ZPPPD preconceptual epigenetic
effects on offspring have become a hot topic. In 2009 Ouko et al at the
University of Witwaterstrand reported on "Effect of alcohol consumption on CpG
methylation in the differentially methylated regions of H19 and IG-DMR in male
gametes: implications for fetal alcohol spectrum disorders":
"Background: Exposure to alcohol in utero is the main
attributable cause of fetal alcohol spectrum disorders (FASD) which in its most
severe form is characterized by irreversible behavioral and cognitive
disability. Paternal preconception drinking is not considered to be a
significant risk factor, even though animal studies have demonstrated that
chronic paternal alcohol consumption has a detrimental effect on the physical
and mental development of offspring even in the absence of in utero alcohol
exposure. It has been documented that alcohol can reduce the levels and activity
of DNA methyltransferases resulting in DNA hypomethylation and that reduced
methyltransferase activity can cause activation of normally silenced genes. The
aim of this study was to establish a link between alcohol use in men and
hypomethylation of paternally imprinted loci in sperm DNA in genomic regions
critical for embryonic development, thus providing a mechanism for paternal
effects in the aetiology of FASD.
"Methods: Sperm DNA from male volunteers was bisulfite
treated and the methylation patterns of 2 differentially methylated regions
(DMRs), H19 and IG-DMR, analyzed following sequencing of individual clones. The
methylation patterns were correlated with the alcohol consumption levels of the
volunteer males.
"Results: There was a pattern of increased
demethylation with alcohol consumption at the 2 imprinted loci with a
significant difference observed at the IG-DMR between the nondrinking and heavy
alcohol consuming groups. Greater inter-individual variation in average
methylation was observed at the H19 DMR and individual clones were more
extensively demethylated than those of the IG-DMR. CpG site #4 in the IG-DMR was
preferentially demethylated among all individuals and along with the H19 DMR CpG
site #7 located within the CTCF binding site 6 showed significant demethylation
in the alcohol consuming groups compared with the control group.
"Conclusion: This study demonstrates a correlation
between chronic alcohol use and demethylation of normally hypermethylated
imprinted regions in sperm DNA. We hypothesize that, should these epigenetic
changes in imprinted genes be transmitted through fertilization, they would
alter the critical gene expression dosages required for normal prenatal
development resulting in offspring with features of FASD."
https://pubmed.ncbi.nlm.nih.gov/19519716/ [3331]
Day et al in "Influence of paternal preconception
exposures on their offspring: through epigenetics to phenotype" (2016) discuss
paternal age (the Defence asserts genetic age supervenes calendar age), paternal
diet (the Defence asserts cannabis and psychedelics improve dietary choices and
that cannabis assists digestive metabolism), environmental toxicants and
psychosocial stressors (the Defence asserts the Town Smell falls into both
categories), and particularly alcohol (the Defence evidence shows cannabis and
psychedelics are associated with reduced alcohol consumption):
"FASDs are a broad array of congenital disorders with
major symptoms including reduced birth weight, impaired cognitive function and
behavior, and neuropsychological deficits in visual-spatial learning. Studies
have shown that paternal alcohol consumption has epigenetic effects on sperm
DNA, suggesting a role in the development of congenital disorders in offspring.
Up to 75% of children with FASD have biological fathers who are alcoholics,
suggesting that preconceptional paternal alcohol consumption negatively impacts
their offspring. It has been shown that teratogens such as alcohol significantly
reduce the activity of DNA methyltransferases, leading to increased CG
hypomethylation and subsequent activation of normally silenced genes. Chronic
paternal alcohol consumption alone hypomethylates his offspring’s genes even in
the absence of maternal alcohol consumption before or during pregnancy. This
epigenetic hypomethylation alters gene expression dosages required for normal
prenatal development, resulting in offspring with characteristic symptoms of
FASDs. Here we examine the effects of paternal alcohol consumption on the
prevalence and symptoms of FASDs and related congenital defects.
"Effects on birthweight and individual organ weights
A hallmark symptom of FASD is decreased newborn birth
weight. Murine studies have shown that offspring from alcohol-treated fathers
have a higher prevalence of low birth weights. A study in rats has shown that
offspring from alcohol-treated fathers decreased in weight by two or more
standard deviations when compared to the average weight of offspring born from
controls. This effect was observed in offspring of both acute and long-term
alcohol-treated fathers, which suggests that epigenetic modifications are
sensitive to even small amounts of paternal alcohol consumption. In addition to
marked decreases in birth weight, studies have shown that alcohol consumption
can alter the weight of individual organs. For example, fathers treated with
alcohol for several weeks prior to breeding were more likely to produce
offspring with increased adrenal weights and decreased spleen weights. This
suggests that paternal alcohol consumption may also have an epigenetic impact on
the gene expression governing individual organ development. Furthermore, autopsy
and brain imaging studies have shown marked reductions in overall brain size,
specifically in the cerebellum, basal ganglia, and corpus callosum. This
observed physiological effect on brain structures can explain impaired cognitive
function displayed by offspring sired by alcohol-treated fathers.
"Effects on cognitive behavior and motor ability
Studies have demonstrated the adverse effects of
paternal alcohol consumption on the cognitive and motor ability of offspring.
For example, one study involved feeding male mice varied liquid alcohol diets
containing 25%, 10%, or 0% ethanol-derived calories (EDC). After 7 to 14 weeks
of diet treatment, the males were bred to non-treated females. Offspring were
then subjected to a forced swim test, in which offspring of alcohol-sired
fathers were more immobile than offspring of fathers receiving 0% EDC. While
this may suggest decreased motor ability due to paternal alcohol consumption,
this seems to also be a species-specific effect. The same study was conducted on
rats, showing opposite results with offspring of alcohol-sired fathers
exhibiting increased mobility.
"One possible explanation for this discrepancy could
be the species’ specific response to stressful situations. In humans, it is
known that children with fetal alcohol syndrome cope poorly with stressful
situations, and therefore display hyperresponsiveness to stress. In these
situations, stressors cause an increased corticosterone response, resulting in
exaggerated reactions to stressful situations. This could explain why the rat
offspring of alcohol-treated fathers exhibited increased mobility when forced to
swim; the hyperresponsiveness to stress may override the impaired motor function
that is normally seen in affected offspring. While these results in mice and
rats may seem paradoxical, the mechanisms being affected need to be isolated and
examined separately to determine causation.
"In addition to this hyperresponsiveness seen in rats,
similar studies have also shown that rats sired by alcohol-treated fathers have
greater difficulty learning new tasks and have impaired spatial learning skills
when subjected to maze tests. Studies also suggest that epigenetic modifications
occur in sperm DNA, which may be passed onto offspring. Further studies need to
be conducted in order to determine the specific mechanism by which these
modifications are passed from father to offspring. Understanding these
mechanisms would provide more insight into preventative measures against FASDs
and similar congenital defects.
"Other epigenetic effects of paternal alcohol
consumption
Paternal alcohol consumption has been implicated in
additional congenital disorders, presumably via epigenetic mechanisms. In an
epidemiological study by the Kaiser Foundation, the frequency and severity of
certain congenital abnormalities were correlated with paternal alcohol
consumption. For example, paternal alcohol consumption was found to be
positively associated with ventricular septal defects in newborn children.
Separate animal studies have also shown that paternal alcohol consumption can
lead to increased susceptibility to Pseudomonas infection. The severity of this
increased susceptibility was found to be identical to that of animals whose
mothers consumed alcohol during pregnancy, suggesting that paternal epigenetic
alterations are as crucial to the development of offspring as maternal ones.
Overall, these studies imply that early changes in a father’s lifestyle can
decrease prevalence of congenital disorders in his offspring."
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4913293/ [3332]
Chang et al (2019) continued this interesting assault
on male exceptionalism with "Preconception paternal alcohol exposure exerts
sex-specific effects on offspring growth and long-term metabolic programming" in
which daddy mice were given 70-days of preconception treatment with alcohol.
Exposed males were then mated to six- to eight-week-old females. At no point
during these experiments were the females ever exposed to the preconception
treatments.
The difficulties of detecting such an effect in humans
are all but insurmountable. Where do you find a sufficient number of children of
sober mothers whose alcoholic father is no longer on the scene, confounding the
nature/nurture analysis? How much did the father drink before the pregnancy? The
Texas A&M University researchers list the references:
"For example, clinical data can correlate paternal
alcoholism with negative impacts on child behavior and cognitive development. In
addition, clinical associations between paternal drinking and increased rates of
congenital abnormalities, as well as decreases in infant birth weight and head
circumference, have been reported. However, in these studies, additional and
often uncontrolled factors such as nutrition, poor housing conditions, maternal
stress, smoking, and parental alcohol use all exert independent effects on child
growth and development. In this setting, it is virtually impossible to identify
a direct link between preconception paternal alcohol use and child development."
However in the mice, with controlled conditions, the
picture was clear:
"Preconception paternal alcohol exposure induced a
prolonged period of fetal gestation and an increased incidence of intrauterine
growth restriction, which affected the male offspring to a greater extent than
the females. While the female offspring of ethanol-exposed males were able to
match the body weights of the controls within the first 2 weeks of postnatal
life, male offspring continued to display an 11% reduction in weight at 5 weeks
of age and a 6% reduction at 8 weeks of age. The observed growth deficits
associated with insulin hypersensitivity in the male offspring, while in
contrast, females displayed a modest lag in their glucose tolerance test. These
metabolic defects were associated with an up-regulation of genes within the
pro-fibrotic TGF-β signaling pathway and increased levels of cellular
hydroxyproline within the livers of the male offspring. We observed suppressed
cytokine profiles within the liver and pancreas of both the male and female
offspring, which correlated with the up-regulation of genes in the
LiverX/RetinoidX/FarnesoidX receptor pathways. However, patterns of gene
expression were highly variable between the offspring of alcohol-exposed sires.
In the adult offspring of alcohol-exposed males, we did not observe any
differences in the allelic expression of Igf2 or any other imprinted genes.
"Conclusions
The impact of paternal alcohol use on child
development is poorly explored and represents a significant gap in our
understanding of the teratogenic effects of ethanol. Our studies implicate
paternal exposure history as an additional and important modifier of
alcohol-induced growth phenotypes and challenge the current maternal-centric
exposure paradigm."
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6341619/ [3328]
2019 continued with "Parental alcohol consumption and
the risk of congenital heart diseases in offspring: An updated systematic review
and meta-analysis" from the Xiangya School of Public Health, Hunan, in which
paternal alcohol was associated with a nearly threefold greater risk than
maternal consumption:
"A total of 55 studies involving 41,747 CHD cases and
297,587 controls were identified. Overall, both maternal (odds ratio (OR) =
1.16; 95% confidence interval (CI): 1.05-1.27) and paternal (OR = 1.44; 95% CI:
1.19-1.74) alcohol exposures were significantly associated with risk of total
CHDs in offspring. Additionally, a nonlinear dose-response relationship between
parental alcohol exposure and risk of total CHDs was observed. With an increase
in parental alcohol consumption, the risk of total CHDs in offspring also
gradually increases. For specific CHD phenotypes, a statistically significant
association was found between maternal alcohol consumption and risk of tetralogy
of fallot (OR = 1.20; 95% CI: 1.08-1.33). Relevant heterogeneity moderators have
been identified by subgroup analysis, and sensitivity analysis yielded
consistent results."
https://academic.oup.com/eurjpc/article-pdf/27/4/410/34258325/eurjpc0410.pdf
[3229]
This was duly reported by the European Society of
Cardiology, who write:
"Drinking alcohol three months before pregnancy (and
during the first trimester for mothers) was associated with a 44% raised risk of
congenital heart disease for fathers and 16% for mothers, compared to not
drinking. Binge drinking, defined as five or more drinks per sitting, was
related to a 52% higher likelihood of these birth defects for men and 16% for
women.
"‘Binge drinking by would-be parents is a high risk
and dangerous behaviour that not only may increase the chance of their baby
being born with a heart defect, but also greatly damages their own health,’ said
study author Dr Jiabi Qin, of Xiangya School of Public Health, Central South
University, Changsha, China.
"Dr Qin said the results suggest that when couples are
trying for a baby, men should not consume alcohol for at least six months before
fertilisation while women should stop alcohol one year before and avoid it while
pregnant.
"Congenital heart diseases are the most common birth
defects, with approximately 1.35 million babies affected every year. These
conditions can increase the likelihood of cardiovascular disease later life,
even after surgical treatment, and are the main cause of perinatal death.
Alcohol is a known teratogen and has been connected with foetal alcohol spectrum
disorder (FASD). Around one in four children with FASD have congenital heart
disease, indicating that alcohol might also be implicated in these disorders.
"Previous studies investigating the link between
alcohol and congenital heart disease have focused on prospective mothers, with
inconclusive results. This is the first meta-analysis to examine the role of
paternal alcohol drinking."
https://www.escardio.org/The-ESC/Press-Office/Press-releases/Fathers-to-be-should-avoid-alcohol-six-months-before-conception
[3330]
Back with the maternal alcohol Flentke et al bring us
up to 2024 with "Alcohol exposure suppresses ribosome biogenesis and causes
nucleolar stress in cranial neural crest cells", in which the progeny of drunk
mummy zebra fish were examined in search of a mechanism involving ribosome
biogenesis, which turned out to be a successful endeavour:
"Prenatal alcohol exposure (PAE) causes cognitive
impairment and a distinctive craniofacial dysmorphology, due in part to
apoptotic losses of the pluripotent cranial neural crest cells (CNCs) that form
facial bones and cartilage. We previously reported that PAE rapidly represses
expression of >70 ribosomal proteins (padj = 10-E47). Ribosome dysbiogenesis
causes nucleolar stress and activates p53-MDM2-mediated apoptosis. Using primary
avian CNCs and the murine CNC line O9-1, we tested whether nucleolar stress and
p53-MDM2 signaling mediates this apoptosis. We further tested whether
haploinsufficiency in genes that govern ribosome biogenesis, using a blocking
morpholino approach, synergizes with alcohol to worsen craniofacial outcomes in
a zebrafish model. In both avian and murine CNCs, pharmacologically relevant
alcohol exposure (20mM, 2hr) causes the dissolution of nucleolar structures and
the loss of rRNA synthesis; this nucleolar stress persisted for 18-24hr. This
was followed by reduced proliferation, stabilization of nuclear p53, and
apoptosis that was prevented by overexpression of MDM2 or dominant-negative p53.
In zebrafish embryos, low-dose alcohol or morpholinos directed against ribosomal
proteins Rpl5a, Rpl11, and Rps3a, the Tcof homolog Nolc1, or mdm2 separately
caused modest craniofacial malformations, whereas these blocking morpholinos
synergized with low-dose alcohol to reduce and even eliminate facial elements.
Similar results were obtained using a small molecule inhibitor of RNA Polymerase
1, CX5461, whereas p53-blocking morpholinos normalized craniofacial outcomes
under high-dose alcohol. Transcriptome analysis affirmed that alcohol suppressed
the expression of >150 genes essential for ribosome biogenesis. We conclude that
alcohol causes the apoptosis of CNCs, at least in part, by suppressing ribosome
biogenesis and invoking a nucleolar stress that initiates their p53-MDM2
mediated apoptosis. We further note that the facial deficits that typify PAE and
some ribosomopathies share features including reduced philtrum, upper lip, and
epicanthal distance, suggesting the facial deficits of PAE represent, in part, a
ribosomopathy."
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0304557
[3333]
2024 also saw a quantification of a threshold dose for
paternal alcohol use in F1 FASD, by Higgins et al at Texas A&M, in "Chronic
paternal alcohol exposures induce dose-dependent changes in offspring
craniofacial shape and symmetry":
"Procrustes ANOVA followed by canonical variant
analysis of geometric facial relationships revealed that Low-, Medium-, and
High-dose treatments each induced distinct changes in craniofacial shape and
symmetry. Our analyses identified a dose threshold between 1.543 and 2.321
g/kg/day. Below this threshold, preconception paternal alcohol exposure induced
changes in facial shape, including a right shift in facial features. In
contrast, above this threshold, paternal exposures caused shifts in both shape
and center, disrupting facial symmetry. Consistent with previous clinical
studies, changes in craniofacial shape predominantly mapped to regions in the
lower portion of the face, including the mandible (lower jaw) and maxilla (upper
jaw). Notably, high-dose exposures also impacted the positioning of the right
eye. Our studies reveal that paternal alcohol use may be an unrecognized factor
contributing to the incidence and severity of alcohol-related craniofacial
defects, complicating diagnostics of fetal alcohol spectrum disorders."
https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2024.1415653/full
[3334]
To put this into context the following represent 10
grams of alcohol.

https://responsibledrinking.eu/what-is-a-standard-drink-of-alcohol/ [3335]
For a 70kg man the threshold for more serious
craniofacial effects in his child would be 108.01 to 162.47 g/day, so this is
between 11 and 16 standard European units.
Alcohol-related epigenetic effects do not stop with
the next generation, as explained in "Epigenetic mechanisms in alcohol- and
adversity-induced
developmental origins of neurobehavioral functioning"
by Boschen et al (2018):
Under "DNA methylation of neurodevelopmental and
plasticity-related genes" we learn:
"Genes related to neurodevelopment and HPA axis
regulation are commonly altered in both developmental stress and alcohol
models."
Under "Expression of epigenetic regulators":
"Another commonality between the consequences of
early-life stress and alcohol are the observed alterations in levels of
epigenetic regulators throughout the brain."
And under "DNA Methylation":
"Brain-derived neurotrophic factor (BDNF) is another
growth factor important for cell proliferation, dendritic outgrowth, and synapse
formation, particularly in the adult brain. Numerous studies have demonstrated
that development alcohol exposure causes long-term changes to functional and
anatomical measures linked to BDNF signaling, including long-term potentiation
(Puglia and Valenzuela, 2010), adult neurogenesis (Boehme et al., 2011;
GilMohapel et al., 2010; Hamilton et al., 2011; Klintsova et al., 2007), and
dendritic morphology (Berman et al., 1996; Boschen et al., 2016; Hamilton et
al., 2015; Hamilton et al., 2010; Redila and Christie, 2006; Whitcher and
Klintsova, 2008). Studies have demonstrated that Bdnf gene expression is altered
following developmental alcohol exposure in a timing and region-specific manner
(Caldwell et al., 2008; Feng et al., 2005; Heaton et al., 2000; Heaton et al.,
2003), though the diverse models and methods of analysis used in these studies
makes it difficult to draw definitive conclusions. Epigenetic modification of
Bdnf has not been well studied in alcohol exposure models, despite being a
significant target of interest in prenatal stress research. Bdnf exon I
hypomethylation was reported 24 h following PD4–9 alcohol exposure in the male
rat hippocampus; this reduction in methylation was correlated with increased
exon I-driven Bdnf gene expression (Boschen et al., 2015). However, by PD72,
methylation status had returned to control levels (Boschen et al., 2016). Bdnf
and other growth factors that could contribute to either delayed embryonic
growth or long-term deficits in neuroplasticity need to be further investigated
from an epigenetic perspective."
Under "Non-coding RNAs":
"Disruption of noncoding RNAs, such as miRNAs, can
cause a wide range of cellular or DNA damage, contributing to long-term deficits
(Figure 1). miRNAs affect the production of protein products through
translational repression or degradation of the mRNA. In vivo and in vitro models
of FASD have identified numerous miRNAs that are impacted by developmental
alcohol exposure, including miR-9, miR-20a, miR-21, miR-30, miR-103, miR-151,
miR-153, miR-335, and miR-140-3p (Balaraman et al., 2014; Balaraman et al.,
2012; Guo et al., 2012; Ignacio et al., 2014; Pappalardo-Carter et al., 2013;
Pietrzykowski et al., 2008; Sathyan et al., 2007; Soares et al., 2012; Tal et
al., 2012; Wang et al., 2009). In a study that examined the effects of prenatal
stress on miRNA expression, dams were administered daily restraint stress and
forced swimming from days 12–18 of gestation (Zucchi et al., 2013). This
stressor disrupted maternal behavior, as reduced tail chasing behavior was
exhibited by dams exposed to stress. In the whole brains of their newborn male
offspring, 336 miRNAs were differentially expressed. Notably, some of the same
miRNAs were altered by prenatal stress as have been mentioned in the
developmental alcohol literature, including miR-9, miR-20a, miR-103, and
miR-151."
"Epigenetic modifications represent an avenue through
which adverse environmental conditions can affect generations beyond the animals
that were directly exposed via intergenerational and transgenerational
transmission. Epigenetic marks can be passed through the paternal germline (F0)
following preconception exposure to stress or alcohol, resulting in
physiological, behavioral, or altered levels of global methylation in the
offspring (F1). Exposure of a fetus (via exposure of the F0 pregnant dam) or
neonate (F1) to alcohol or stress affects not only the directly exposed animal
but also the developing germline. Altered epigenetic marks can then be
transmitted to animals that were never exposed to the stressor or alcohol in the
F2 generation and beyond, resulting in changes to methylation associated with
multiple genes and epigenetic regulators and ppersistent behavioral
perturbations. AE: Alcohol exposure; ELS: Early life stress."
https://europepmc.org/backend/ptpmcrender.fcgi?accid=PMC5856624&blobtype=pdf
[3337]
Portugalov and Akirav (2024) have the latest on the
FAAH blocking approach with "FAAH Inhibition Reverses Depressive-like Behavior
and Sex-Specific Neuroinflammatory Alterations Induced by Early Life Stress":
"Early life stress (ELS) increases predisposition to
major depressive disorder (MDD), with neuroinflammation playing a crucial role.
This study investigated the long-term effects of the fatty acid amide hydrolase
(FAAH) inhibitor URB597 on ELS-induced depressive-like behavior and messenger
RNA (mRNA) of pro-inflammatory cytokines in the medial prefrontal cortex (mPFC)
and CA1 regions. We also assessed whether these gene expression alterations were
present at the onset of URB597 treatment during late adolescence. ELS induced a
depressive-like phenotype in adult male and female rats, which was reversed by
URB597. In the mPFC, ELS downregulated nuclear factor kappa B1 (nfκb1) in both
sexes, while URB597 normalized this expression exclusively in males. In females,
ELS downregulated interleukin (il) 6 and tumor necrosis factor alpha (tnfα) but
upregulated il1β and corticotropin-releasing factor (crf); URB597 normalized
il6, il1β, and crf. In the CA1, ELS downregulated il1β and tnfα in males and
upregulated il1β expression in females, which was reversed by URB597. Some of
these effects began in late adolescence, including mPFC-nfκb1 expression in both
sexes, mPFC-il6 and mPFC-il1β in females, CA1-il1β and CA1-tnfα in males, and
CA1-il1β in females. These findings highlight URB597 as a therapeutic approach
for reversing ELS-induced depressive-like behavior by associating with changes
in the gene expression of neuroinflammatory cytokines, with notable sex
differences."
https://www.mdpi.com/2073-4409/13/22/1881 [3993]
The discrimination of the ZPPPD against subjects of
early life stress as overproducers of FAAH is further demonstrated by Demaili et
al (2023) in "Epigenetic (re)programming of gene expression changes of CB1R and
FAAH in the medial prefrontal cortex in response to early life and adolescence
stress exposure":
"Thus, the present study addressed the hypotheses that
ELS and adolescent stress differentially affect the expression of regulatory
elements of the endocannabinoid system, cannabinoid receptor 1 (CB1R) and fatty
acid amide hydrolase (FAAH) in the medial prefrontal cortex (mPFC) of adult
female rats. We also tested the hypothesis that the proposed gene expression
changes are epigenetically modulated via altered DNA-methylation. The specific
aims were to investigate if (i) ELS and adolescent stress as single stressors
induce changes in CB1R and FAAH expression (ii) ELS exposure influences the
effect of adolescent stress on CB1R and FAAH expression, and (iii) if the
proposed gene expression changes are paralleled by changes of DNA methylation.
The following experimental groups were investigated: (1) non-stressed controls
(CON), (2) ELS exposure (ELS), (3) adolescent stress exposure (forced swimming;
FS), (4) ELS + FS exposure. We found an up-regulation of CB1R expression in both
single-stressor groups and a reduction back to control levels in the ELS + FS
group. An up-regulation of FAAH expression was found only in the FS group. The
data indicate that ELS, i.e., stress during a very immature stage of brain
development, exerts a buffering programming effect on gene expression changes
induced by adolescent stress. The detected gene expression changes were
accompanied by altered DNA methylation patterns in the promoter region of these
genes, specifically, a negative correlation of mean CB1R DNA methylation with
gene expression was found. Our results also indicate that ELS induces a
long-term “(re)programming” effect, characterized by CpG-site specific changes
within the promoter regions of the two genes that influence gene expression
changes in response to FS at adolescence."
https://www.frontiersin.org/journals/cellular-neuroscience/articles/10.3389/fncel.2023.1129946/full
[3994]
Meanwhile, according to Astley et al (1992):
"The association between fetal marijuana and/or
alcohol exposure and facial features resembling fetal alcohol syndrome was
investigated in a sample of 80 children. Standardized lateral and frontal facial
photographs were taken of 40 children, 5 to 7 years of age, whose mothers
reported frequent use of marijuana during the first trimester of pregnancy and
40 children whose mothers reported no use of marijuana during pregnancy. The
marijuana-exposed and unexposed children were group-matched on alcohol exposure
prior to and during pregnancy, sex, race, and age at the time of assessment. The
photographs were assessed clinically by a study staff dysmorphologist and
morphometrically by computerized landmark analysis. Fetal alcohol syndrome-like
facial features were not associated with prenatal marijuana exposure in this
study sample."
https://depts.washington.edu/fasdpn/pdfs/astley-1992.pdf [3336]
No rescheduling of alcohol or changes have been made
to the ZPPPD in the light of these findings about ugliness.
I am obliged to the 28 February 2024 BBC article "Why
alcohol is so dangerous for young adults' brains":
"In the past, neural development was thought to stop
in our early teens, but a swathe of recent research shows that the adolescent
brain undergoes a complex rewiring that does not end until at least the age of
25.
"The most important changes include a decline in 'grey
matter' as the brain prunes away the synapses that allow one cell to communicate
with another. At the same time, white matter – long-distance connections known
as axons covered with an insulating fatty sheath – tends to proliferate. 'They
are like the brain's super-highways,' says Lindsay Squeglia, a neuropsychologist
at the Medical University of South Carolina. The result is a more efficient
neural network that can process information more quickly.
"The limbic system, involved with pleasure and reward,
is the first to mature. 'These areas are fully adult-like during adolescence,'
Squeglia explains. The prefrontal cortex, which is located behind the forehead,
is slower to ripen. This region is responsible for higher-order thinking – which
includes emotional regulation, decision-making, and self-control.
"The relative imbalance of these two regions'
development can explain why adolescents and young adults tend to be more
risk-taking than adults. 'A lot of people describe the adolescent brain as
having a fully developed gas pedal without brakes,' says Squeglia. And bathing
our neurons in alcohol – which is known to release inhibition – may only amplify
this thrill chasing. For particularly impetuous teenagers, alcohol can create a
vicious cycle of bad behaviour and delinquency. 'The more impulsive kids tend to
drink more, and then drinking causes more impulsivity,' says Squeglia.
"At high enough frequencies and volumes, adolescent
drinking could impair the brain's long-term development. Longitudinal studies
show that early drinking is associated with a more rapid decline in grey matter,
while the growth of the white matter is stunted. 'Those super-highways aren't
getting paved as much in kids who start drinking,' says Squeglia.
"The consequences may not be immediately evident in
cognitive tests; in a young brain, the regions responsible for problem solving
can work a little bit harder to make up for the deficits. It cannot keep this up
forever, however. 'After multiple years of drinking, we see less activation in
the brain and poorer performance on these tests,' says Squeglia.
"Early drinking can also take its toll on mental
health, and heightens the risk of alcohol abuse later in life. This is
particularly true for people who have a family history of alcoholism – the
earlier they start, the greater their chances of developing a drinking problem
themselves. The genes associated with an advanced risk of alcohol abuse seem to
be most influential during this critical period of brain development. 'And the
longer that someone is able to wait, the less likely these genes are going to
come into play,' says Squeglia."
https://www.bbc.com/future/article/20240228-how-alcohol-affects-teens-and-young-adults-brains
[4474]
But how many schizophrenics have a concrete reason to
suspect their genetic susceptibility?
It's not as if doctors in Slovenia are offering
genetic screening to see whether patients are likely to benefit or otherwise
from THC.
For as the authors say,
"The widespread use of cannabis calls for a concerted
effort into increased understanding of both the positive and negative effects of
the drug."
But it seems they do not agree with themselves, for
when it comes to Figure 3
https://www.nature.com/articles/s41398-018-0137-3 [1899]
In 2010 it emerged from a study of brain volumes in
schizophrenics and non-schizophrenics tagged for the common functional
polymorphism, Val108/158Met:
"Across patients and controls, each copy of the COMT
met allele was associated on average with a 2.6% increase in right amygdala
volume, a 3.8% increase in left amygdala volume and a 2.2% increase in right
hippocampus volume. There were no effects of COMT genotype on volumes of the
whole brain and prefrontal regions.
"Thus, the COMT Val108/158Met polymorphism was shown
to influence medial temporal lobe volumes in a linear-additive manner, mirroring
its effect on dopamine catabolism."
https://www.sciencedirect.com/science/article/abs/pii/S1053811909013354
[213]
However a 2018 attempt to link COMT variability to
cannabis use in psychotic cases was "unconvincing".
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5812637/ [214]
The results of Cobb
Scott et al (2019) were fairly uneventful, even in a sample with higher than
usual levels of alcohol use and psychopathology:
"Frequent cannabis use during adolescence has been
associated with alterations in brain structure. However, studies have featured
relatively inconsistent results, predominantly from small samples, and few
studies have examined less frequent users to shed light on potential brain
structure differences across levels of cannabis use. In this study,
high-resolution T1-weighted MRIs were obtained from 781 youth aged 14–22 years
who were studied as part of the Philadelphia Neurodevelopmental Cohort. This
sample included 147 cannabis users (109 occasional [≤1–2 times per week] and 38
frequent [≥3 times per week] users) and 634 cannabis non-users. Several
structural neuroimaging measures were examined in whole brain analyses,
including gray and white matter volumes, cortical thickness, and gray matter
density. Established procedures for stringent quality control were conducted,
and two automated neuroimaging software processing packages were used to ensure
robustness of results. There were no significant differences by cannabis group
in global or regional brain volumes, cortical thickness, or gray matter density,
and no significant group by age interactions were found. Follow-up analyses
indicated that values of structural neuroimaging measures by cannabis group were
similar across regions, and any differences among groups were likely of a small
magnitude. In sum, structural brain metrics were largely similar among
adolescent and young adult cannabis users and non-users. Our data converge with
prior large-scale studies suggesting small or limited associations between
cannabis use and structural brain measures in youth. Detailed studies of
vulnerability to structural brain alterations and longitudinal studies examining
long-term risk are clearly indicated."
But noting that:
"Previous studies with small samples were likely underpowered to detect small
magnitude effects, which could partially explain variability in the literature.
Our results converge with data from larger samples of cannabis-using youth,
which have found more limited brain structural differences associated with
cannabis than smaller studies. For example, Weiland and colleagues compared 50
adolescent daily users of cannabis to 50 demographically matched non-users,
replicating methods from an earlier study, and found non-significant differences
in volume, surface-based morphometry, and shape. Similarly, in a sample of 439
adolescents, Thayer and colleagues found no significant associations of past
month cannabis use with brain volume or measures of diffusivity after covarying
for alcohol use disorder symptoms. Our results extend these studies by probing
effects across levels of cannabis use and examining measures of cortical
thickness and gray matter density."
https://www.nature.com/articles/s41386-019-0347-2 [5401]
In the worrying-sounding "Grey Matter Volume
Differences Associated with Extremely Low Levels of Cannabis Use in Adolescence"
from Orr et al (2019) it turns out that:
"Preclinical evidence has consistently demonstrated a
causal relationship between cannabis exposure and changes to brain morphology
(for review, see Panlilio and Justinova, 2018). The human evidence, however, has
been variable reporting both increases and decreases in brain volumes (Ashtari
et al., 2011; Cousijn et al., 2012; Gilman et al., 2014), no volume differences
(Jager et al., 2007; Weiland et al., 2015; Gillespie et al., 2018), and modest
effect sizes (Weiland et al., 2015). Factors including the age of cannabis use
initiation, comorbid substance use, and levels of use are believed to contribute
to variability in the human findings (Curran et al., 2016)."
...
"One study has reported differences in gray-matter
density and shape of the amygdala and nucleus accumbens in recreational cannabis
users (Gilman et al., 2014), but subsequent research has suggested that these
findings may be associated with alcohol (Weiland et al., 2015) and nicotine
(Gillespie et al., 2018) exposure in the cannabis users."
...
"The eCB system mediates maturation-related neural
reorganization (Fernández-Ruiz et al., 2000), which may place adolescents at
heightened vulnerability to structural brain effects of cannabis exposure as
adolescence is a time of rapid neural maturation (Rubino and Parolaro, 2008).
Consistent with this suggestion, those who commenced cannabis use in adolescence
typically show greater structural brain differences than those who initiated use
in adulthood (Battistella et al., 2014; Lubman et al., 2015). These findings may
also have been influenced by the effects of other substances, however, as one
study comparing adolescent daily cannabis users with controls matched for
alcohol and nicotine use found no differences in subcortical gray-matter density
or morphology (Weiland et al., 2015)."
...
"It is possible that cannabis, alcohol, and nicotine
have differential effects on brain morphometry; specifically, recreational
cannabis use has been associated with volume increases, whereas alcohol has been
associated with volume reductions. In the current study, we matched the groups
on alcohol and nicotine use and, within the cannabis using group, neither
alcohol nor nicotine use was associated with individual differences in GMV,
suggesting that the GMV differences we report are associated with cannabis use."
https://www.jneurosci.org/content/39/10/1817 [3721]
Slovenia has not proposed banning alcohol on the
grounds of reduced brain volume. Of course it may be that Slovenians have a
religious belief in alcohol and reduced brain volume. So they consider
brain-expanding cannabis, even at extremely low levels, to be heretical and a
sin, considering their prior inaccurate impression that cannabis was more, and
not 114 times less [852],
harmful than alcohol, if we exclude prohibition effects and the sin. The Court
is invited to compare the constitutionality of exercising these opposing
beliefs.
Even one or two units of alcohol per day reduce brain
volume, isn't that true?
"...the associations between alcohol intake and brain
structure using multimodal imaging data from 36,678 generally healthy
middle-aged and older adults from the UK Biobank, controlling for numerous
potential confounds. Consistent with prior literature, we find negative
associations between alcohol intake and brain macrostructure and microstructure.
Specifically, alcohol intake is negatively associated with global brain volume
measures, regional gray matter volumes, and white matter microstructure. Here,
we show that the negative associations between alcohol intake and brain
macrostructure and microstructure are already apparent in individuals consuming
an average of only one to two daily alcohol units, and become stronger as
alcohol intake increases."
Reagan Wetherill, PhD, of the University of
Pennsylvania Perelman School of Medicine in Philadelphia and co-authors report
an increase from two alcohol units to three showed changes equivalent to aging
3.5 years, and
"A recent meta-analysis of individuals with AUD
(n = 433) showed lower gray matter volume (GMV) in the corticostriatal-limbic
circuits, including regions of the prefrontal cortex, insula, superior temporal
gyrus, striatum, thalamus, and hippocampus compared to healthy controls
(n = 498). Notably, lower GMV in striatal, frontal, and thalamic regions was
associated with AUD duration or lifetime alcohol consumption. Although alcohol
consumption can produce global and regional tissue volume changes, frontal
regions are particularly associated with these effects. Further, research
suggests that the effects of alcohol consumption on brain volume interact with
the effects of aging."
https://www.nature.com/articles/s41467-022-28735-5 [1049]
On the other hand "Cannabis dose and age of onset of
cannabis use did not affect hippocampal volumes", according to a 2017 study of
20 "heavy cannbis users" by Koenders et al with an average 39 month follow-up.
https://pmc.ncbi.nlm.nih.gov/articles/PMC5544121/ [3724]
Any study of brain volume which disregards the major
confounder tobacco is seriously compromised. Daniju et al (2022) found:
"Regular cannabis users who also smoked tobacco
cigarettes showed altered GMV patterns relative to controls. However, a similar
pattern of GMV differences was also seen between regular tobacco users that did
not use cannabis. Further research is needed to disentangle the effects of
cannabis and tobacco use on brain structure."
In other words, they didn't get the anti-cannabis
result they were hoping for, but they managed not to look at any cannabis users
who did not use tobacco.
https://pmc.ncbi.nlm.nih.gov/articles/PMC9716493/ [3725]
When Snoop Dogg posted this photo comparison with
tragic drunk English footballer Paul Gascoigne...

...Gascoigne responded by challenging the rapper to a
12-round boxing match. So this is not a scientific debate.
Another cause of brain shrinkage you won't hear much
about is antipsychotics.
In 2015 Vita et al reported:
"Longitudinal magnetic resonance imaging studies
comparing changes in the volume of cortical GM over time between patients with
schizophrenia and healthy control subjects published between January 1, 1983,
and March 31, 2014, were analyzed. Hedges' g was calculated for each study and
volume changes from baseline to follow-up were analyzed. Meta-regression
statistics were applied to investigate the role of potential moderators of the
effect sizes.
"Results: Eighteen studies involving 1155 patients
with schizophrenia and 911 healthy control subjects were included. Over time,
patients with schizophrenia showed a significantly higher loss of total cortical
GM volume. This was related to cumulative antipsychotic intake during the
interval between scans in the whole study sample. Subgroup meta-analyses of
studies on patients treated with second-generation antipsychotics and
first-generation antipsychotics revealed a different and contrasting moderating
role of medication intake on cortical GM changes: more progressive GM loss
correlated with higher mean daily antipsychotic intake in patients treated with
at least one first-generation antipsychotic and less progressive GM loss with
higher mean daily antipsychotic intake in patients treated only with
second-generation antipsychotics."
https://pubmed.ncbi.nlm.nih.gov/25802081/ [2161]
There are no laws against coffee are there?
You don't need a prescription to buy coffee?
There are no legal controls on how much coffee you can
drink, are there?
There are no roadside caffeine tests, are there?
And yet too much coffee is bad for you, would you
agree?
According to the Independent March 3 2022:
"A father died after accidentally downing caffeine
powder as strong as 200 cups of coffee, an inquest heard.
"Personal trainer Thomas Mansfield, 29, ordered a 100g
packet of caffeine powder to make supplement drinks at his family home.
"But he accidentally made a mixture containing seven
times the recommended dose before he "necked" it.
and
"A post mortem found the caffeine per litre of blood
in his system was the equivalent of up to 200 cups of coffee.
"The inquest heard that death could be caused by
significantly lower levels than the amount he consumed."
His medical cause of death was given as caffeine
toxicity.
https://www.independent.co.uk/news/uk/home-news/personal-trainer-dead-caffeine-drink-b2026650.html
[1056]
So with this limited human sample of 1 that puts
caffeine in the vicinity of nitrous oxide and at least five times more dangerous
than psilocybin, LSD and marijuana, referring to:
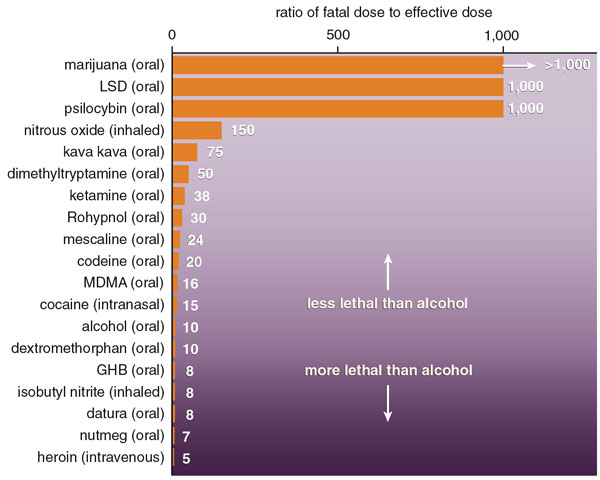
There were no calls for this product to be banned.
There were no murder or manslaughter charges to anyone who supplied or assisted
him?
Because we know that makes no sense, and would only
makes things worse.
But this case did not concern actual coffee, and we
can barely imagine consuming 200 cups of coffee.
What about chronic use?
There were inverse linear associations between
habitual coffee consumption and total brain (fully adjusted β per cup −1.42, 95%
CI −1.89, −0.94), grey matter (β −0.91, 95% CI −1.20, −0.62), white matter (β
−0.51, 95% CI −0.83, −0.19) and hippocampal volumes (β −0.01, 95% CI −0.02,
−0.003), but no evidence to support an association with white matter
hyperintensity (WMH) volume (β −0.01, 95% CI −0.07, 0.05). The association
between coffee consumption and dementia was non-linear
(Pnon-linearity = 0.0001), with evidence for higher odds for non-coffee and
decaffeinated coffee drinkers and those drinking >6 cups/day, compared to light
coffee drinkers. After full covariate adjustment, consumption of >6 cups/day was
associated with 53% higher odds of dementia compared to consumption of 1–2
cups/day (fully adjusted OR 1.53, 95% CI 1.28, 1.83), with less evidence for an
association with stroke (OR 1.17, 95% CI 1.00, 1.37, p = 0.055).
https://www.tandfonline.com/doi/abs/10.1080/1028415X.2021.1945858 [215]
[1-β = probability of a "true positive", i.e.,
correctly rejecting the null hypothesis]
Do you think a campaign to ban coffee in Slovenia on
health grounds would have much public support or political traction?
You would probably agree that coffee brought from the
Near East was already established in eastern Europe by the 17th century?
Let's go back as far as we can in the drug war over
coffee to see what parallels we can find.
"The Greeks claim that a Greek opened the first
coffeehouse in Constantinople in 1475 at a place called Kiva Han, possibly in
today's Tahtakale district, although Turkish sources suggest coffee was only
brought to the city in 1517 after Sultan Selim I (r. 1512-1520) conquered Egypt.
Yet another story relates how the Ottoman governor of Yemen, Özdemir Paşa, had
learned to love coffee while there and brought coffee beans with him when he was
called to Constantinople where he introduced coffee drinking to Kanuni Sultan
Süleyman (r. 1520-1566)."
Prohibition had little effect: the only trouble seemed
to come from religious people:
"A dispute broke out in Constantinople, as it had
earlier in Cairo, about drinking coffee. Purist Muslims wanted it banned because
of its stimulating nature and because it was an innovation. The chief religious
authority among the Ottomans during the reign of Süleyman and his successor,
Şeyhülislam Ebussuud issued a fatwa, or decree, against drinking coffee. The
year was 1543, according to Refik Ahmet Sevengil, and ships had arrived bearing
cargo of coffee beans. As a result of the fatwa, the bags of beans were dumped
in the sea. Over the years, however, the prohibition was mostly ignored and the
first coffeehouse opened in Istanbul in 1555, according to Ottoman sources. And
when Bostanzade Mehmed Efendi became şeyhülislam toward the end of the 16th
century, he issued another fatwa, in poetic form no less, indicating that
suspicions about coffee were groundless."
https://www.hurriyetdailynews.com/coffee-and-coffeehouses-among-the-ottomans-76123
[892]
According to "Coffee and the Ottoman Social Sphere" by
Marita Ervin (2014):
"The most common and perhaps most accurate explanation
accepted by historians today is that coffee originated from the Sufi orders of
Yemen where caffeine was essential to religious practice. Sufis were an
untraditional sect of Islam whose methods were seen as erratic and profane to
both the Sunnis and Shiites. As opposed to practicing the structured prayer
indicative of traditional Islamic sects, the Sufis consumed narcotics and danced
fitfully to demonstrate their devotion to Allah. Such a practice required the
consumption of a drug that could sustain them for many days. The Sufis lived on
the fringes of Ottoman society and coffee's connection to their orders stirred
speculation. Kâtib Celebi, the famous Ottoman historian, described coffee's
first users as 'Certain sheiks, who lived with their dervishes in the mountains
of Yemen, used to crush and eat their berries, which they called qalb wabūn, […]
a cold dry food, suited to the ascetic life and sedative of lust.' Celebi noted
coffee's connection with social outcasts and the need to repress the desire to
sin. Although Ottoman coffee struggled with its Sufi association, the Sufi
concept of developing a bond over a cup of coffee remained an inherent quality
of the beverage. Two men recognized the commercial potential of coffee's
cohesive abilities and harnessed it into the development of the coffeehouse.
After the coffee plant was extracted from the Sufi lodges and brought to
Istanbul by traders, two Syrians merchants named Hakam and Shams opened the
first coffeehouse in the Ottoman capital in 1555."
For a time things were a roaring success:
"Swedish traveler Mouradgea D'Ohsson described over
six-hundred coffeehouses existing in Istanbul in the 1570s, only fifteen years
after coffee first arrived in Istanbul."
"Englishman George Sandys explained that the Turks
enjoyed their coffee 'as hot as they can
suffer it: black as soote, and tasting not much unlike
it.'"
"Within the coffeehouse patrons were immersed in a
safe atmosphere that allowed them to engage in a means of creative self
expression through poetry. When poetry was not being recited urban storytellers
recounted traditional Ottoman tales and sophisticated stories that catered to an
academic audience. Customers were able to play games such as chess and
backgammon that involved strategy without the haram gabling component. This wide
range of stimulating coffeehouse activities were not confined to a specific
class. Ottoman historian Ibramhimi Pechevi reported that there was no typical
coffeehouse customer outside of the fact that they were only Muslims and
predominantly male. He explained:
"'It reached such a point that all kinds of unemployed
officers, judges and professors all seeking preferment, and corner-sitters with
nothing to do proclaimed that there was no place like [the coffeehouse] for
pleasure and relaxation, and filled it until there was no room to sit or stand.
It became so famous that, besides the holders of high offices, even great men
could not refrain from coming there.'"
https://soundideas.pugetsound.edu/cgi/viewcontent.cgi?article=1007&context=history_theses
[891]
The Coffea arabica plant is native to the Caffa
region, in Abyssinia, from which it arrived in Yemen and subsequently to Mecca
and Medina, where it took the name of Qahvaè.
In the Middle Ages, almost nothing was known about
coffee in the West; this aroma made its appearance in Europe in the sixteenth
century, and its widespread diffusion dates back to the defeat of the Turkish
troops, in siege of Vienna in 1683, followed in 1700 by its introduction into
South America.
People liked it.
But you'll know that some reacted with suspicion and
fear, calling the black beverage "amara invenzione di Satana"?
You'll know that they kicked up a religious debate
about the sinfulness of coffee users or, as we would characterise it today,
"coffee use disorder" and "coffee trafficking" were "crimes against society".
Coffee, we could point out, can make certain
susceptible groups anxious or jittery?
We could say it increases heart rate?
And it is diuretic?
We could warn the public that caffeine blocks
adenosine receptors?
We could warn the public that caffeine interferes with
sleep cycles?
Could we reasonably say coffee is a drug which creates
a tolerance?
Would we be lying if we said there are some withdrawal
symptoms?
And yet we can find both responsible and irresponsible
users?
And do we say coffee is like heroin.
So coffee, which we have decided is not a drug in
legal parlance, is a two-edged sword according to these drug criteria. If we
care about addiction, health effects and moral character coffee is both good and
bad. It is hard to draw a line.
And we assume we have a situation where the law
reflects what we know about such dangers.
Perhaps you know how this legal situation came to a
head?
Do you happen to now which Pope was asked to decide on
this question?
Clement VIII.
And do you know the outcome?
He liked it, so it was alright for everyone to like
it. Besides which his friends was a-gonna make a lorra lorra money!
No fan of clemency, Clement executed and plundered
from the get-go. Menocchio, a miller who had found time to come up with the idea
that God was not eternal but had Himself once been created out of chaos, and of
panpsychist Giordano Bruno, a wandering intellectual (and whose cosmopolitan
views had taken him to London and Paris at a dangerous time for nonconformists.
In England Bruno was liked by Henry VIII, may have
spied on the Catholic conspirators for Walsingham, and was mocked by the
Archbishop of Canterbury-to-be George Abbot for supporting "the opinion of
Copernicus that the earth did go round, and the heavens did stand still; whereas
in truth it was his own head which rather did run round, and his brains did not
stand still".
Bruno was imprisoned in Rome for a trial lasting seven
years as the charges were extended to include:
"holding opinions contrary to the Catholic faith and
speaking against it and its ministers;
holding opinions contrary to the Catholic faith about
the Trinity, divinity of Christ, and Incarnation;
holding opinions contrary to the Catholic faith
pertaining to Jesus as the Christ;
holding opinions contrary to the Catholic faith
regarding the virginity of Mary, mother of Jesus;
holding opinions contrary to the Catholic faith about
both Transubstantiation and the Mass;
claiming the existence of a plurality of worlds and
their eternity;
believing in metempsychosis and in the transmigration
of the human soul into brutes;
dealing in magics and divination" (Bruno was an expert
in mnemonics and did memory tricks).
Pope Clement took part in the trial personally.
Bruno's parting shot to the judges:
"Maiori forsan cum timore sententiam in me fertis quam
ego accipiam ("Perhaps you pronounce this sentence against me with greater fear
than I receive it").
"He was turned over to the secular authorities. On 17
February 1600, in the Campo de' Fiori (a central Roman market square), with his
'tongue imprisoned because of his wicked words', he was hung upside down naked
before finally being burned at the stake."
You might think that in an atmosphere like this
coffee, from the Muslim Arab world, would stand little chance. There may even be
people in Ptuj today who don't drink coffee because it is too foreign and
Islamic.
"When coffee first reached Rome, Christian priests
believed that Satan had invented coffee as a substitute for wine which Muslims
were not allowed to drink. Since wine was used in Christian practices such as
Holy Communion, priests thought that coffee must then be from the Anti-Christ.
Faced with strong beliefs that coffee was the drink of Satan, Pope Clement VIII
asked to try a cup before making a decision. When he did, he blessed the drink
as a Christian beverage, resulting in massive imports of coffee to Italy and the
Western world."
https://web.archive.org/web/20100601194012/http://www.professorshouse.com/food-beverage/beverages/coffee-facts-statistics.aspx
[882]
What an excellent idea. We should get (head of UNODC)
Ghada Fathi Waly to smoke some weed, maybe on a different day take a trip, so we
can all receive her seal of approval and relax.
And yet over in Constantinople, forty years after
Bruno's execution in 1600, with the accession of Ibrahim I the Ottoman attitudes
to coffee were softened slightly, but you were still at risk of execution if
caught drinking coffee. His Duterte-like predecessor Marud IV would stalk the
lowest taverns with a hundred pound broadsword and decapitate offenders on the
spot, besides using random people for target practice with his bow and arrow.
Ibrahim was more of a bag in the river man, with 280 concubines drowned, besides
the drug users. Eventually he made such a mess of things the regime was in
trouble, and even his own mother approved his execution, the Ottomans' second
regicide.
https://en.wikipedia.org/wiki/Ibrahim_of_the_Ottoman_Empire#Deposition_and_execution
[883]
"According to one story, an Ottoman Grand Vizier
[believed to be Köprülü, 892] secretly visited a coffeehouse in Istanbul.
"He observed that the people drinking alcohol would
just get drunk and sing and be jolly, whereas the people drinking coffee
remained sober and plotted against the government," says [author of The Devil's
Cup: Coffee, the Driving Force in History, Stewart] Allen."
https://www.npr.org/sections/thesalt/2012/01/10/144988133/drink-coffee-off-with-your-head
[884]
According to the French orientalist Antoine Galland,
"to his great dismay, 'he heard grave people
discoursing seriously on the affairs of the empire, blaming the ministry, and
deciding matters of the utmost importance. He likewise went into the taverns,
where he saw people singing, or talking only of their amours or exploits in
battle, being for the most part soldiers, whom he did not think it convenient to
deprive of this amusement.'' But he had the coffeehouses closed for a time.'" [892]
All of which brings us to the germane question in this
trial, which is whether there is any difference between the sixteenth and
twenty-first century legal doctrine and procedure on the question of principle
whether the formulation of philosophical or religious or political opinions
supposedly contrary to orthodoxy provides sufficient basis for the prosecution,
trial, sentencing or execution of a thinker.
It's not the what, but the why. Over and over we see
the same pattern repeated: an establishment looking for enemies, a novel drug
culture with anti-establishment ideas, a prohibition widely ignored, jealous
rivals e.g. mosques where "imams and expounders of the law were left to keep
company with their beards" [892],
a disproportionate crackdown with opportunities for plunder, and the risk of
potentially fatal consequences from the authorities.
For would the public
health have been improved if Pope Clement had hated coffee, or refused to
experience it or listen to its supporters? He did not know what he did not know.
The situation for coffee users ever since would be analogous to the situation of
the Defendant, while a certain proportion of the population would be deprived of
legal access to life-extending goods, as described in
"The
impact of coffee subtypes on incident cardiovascular disease, arrhythmias, and
mortality: long-term outcomes from the UK Biobank"
by
Chieng
et al (2022):
"Coffee subtypes were
defined as decaffeinated, ground, and instant, then divided into 0, <1, 1, 2–3,
4–5, and >5 cups/day, and compared with non-drinkers. Cardiovascular disease
included coronary heart disease, cardiac failure, and ischaemic stroke. Cox
regression modelling with hazard ratios (HRs) assessed associations with
incident arrhythmia, CVD, and mortality. Outcomes were determined through ICD
codes and death records. A total of 449 563 participants (median 58 years, 55.3%
females) were followed over 12.5 ± 0.7 years. Ground and instant coffee
consumption was associated with a significant reduction in arrhythmia at 1–5
cups/day but not for decaffeinated coffee. The lowest risk was 4–5 cups/day for
ground coffee [HR 0.83, confidence interval (CI) 0.76–0.91, P < 0.0001] and 2–3
cups/day for instant coffee (HR 0.88, CI 0.85–0.92, P < 0.0001). All coffee
subtypes were associated with a reduction in incident CVD (the lowest risk was
2–3 cups/day for decaffeinated, P = 0.0093; ground, P < 0.0001; and instant
coffee, P < 0.0001) vs. non-drinkers. All-cause mortality was significantly
reduced for all coffee subtypes, with the greatest risk reduction seen with 2–3
cups/day for decaffeinated (HR 0.86, CI 0.81–0.91, P < 0.0001); ground (HR 0.73,
CI 0.69–0.78, P < 0.0001); and instant coffee (HR 0.89, CI 0.86–0.93, P <
0.0001).
"Conclusion
Decaffeinated, ground, and instant coffee, particularly at 2–3 cups/day, were
associated with significant reductions in incident CVD and mortality. Ground and
instant but not decaffeinated coffee was associated with reduced arrhythmia."
https://academic.oup.com/eurjpc/article/29/17/2240/6704995?login=false
[5043]
Drug interdiction is one method by which rule by
strength in a chaotic society comes to be legally somatized. But even
incomplete, the cure for a largely imaginary disease is more immoral than the
morally neutral problem, hellishly portrayed by supporters of prohibition.
The source of the word "addiction" comes from the
Latin addictus: in Roman society you could be assigned to someone as a slave if
you could not pay a debt. America's jails have sent marijuana prisoners by the
thousand to the chicken factory. What morality is this?
Being black and disproportionately arrested already,
the American prisoner is no less a slave than his Roman forbear, and this
poultry slavery is the successor to cotton slavery:
"Cotton production dominated the [Georgia] region up
until the postwar period. During this time, poultry production remained small
scale, localized, and was considered "women's work," a common practice among
white and Black households. Yet, in the crisis of the Great Depression, white
landowning farmers and merchants in Northeast Georgia took over the industry and
its profits. This takeover was structured along the preexisting crop lien system
used in the overproduction of cotton that previously dominated the region.
Racially discriminatory state interventions under the Agricultural Adjustment
Act worked together with an agricultural credit system and the consolidation of
white-only farmer cooperatives to create structural barriers to commercial
poultry production for Black and poor white sharecroppers and tenant farmers.
These programs subsidized cotton farmers to idle land and displace farm labor.
In some cases, this quite literally meant replacing the tenant house with the
chicken house.
"Cotton capital, built on the labor of enslaved people
in the South, was mobilized directly and indirectly into the growing poultry
industry, with cotton farmer cooperatives collectively shifting to poultry
production. After this initial takeoff, the Second World War offered up a second
major boost in 1944 when the War Food Administration reserved all the chicken
produced from seven counties in north Georgia. This guaranteed capital allowed
new forms of vertical integration, led by poultry industry innovators John W.
Tyson, Jesse Jewell, and D. W. Brooks. This model of ownership, reliant on
bigger farms and highly industrialized agriculture, restructured the rural South
for the creation of cheap food. Cotton's transformation, coupled with poultry's
takeoff, enabled the 'selling of the South' primed for low-wage, antilabor
industrial growth. The poultry industry thrived off the movement of farmworkers
into processing plants. Organized abandonment produced the poultry capital of
the world through racialized forms of dispossession and displacement,
exclusionary federal loans, purchasing guarantees, and private industry
integration at home, bolstered through war imperialism abroad."
https://monthlyreview.org/2020/07/01/poultry-and-prisons/ [886]
Agro-slavery had continued after the Civil War in
America:
"While the 13th Amendment abolished slavery and
involuntary servitude, it carved out a loophole that allowed for the
exploitation of incarcerated people, who were then and now, disproportionately
Black.
"The amendment abolished slavery and involuntary,
'except as punishment for crime whereof the party shall have been duly
convicted.' Prisoners — men, women, and hundreds of children as young as 6 or 7
— were then leased to private farmers and business owners who'd previously
depended on cheap labor supplied by slaves."
"The 13th Amendment continues to permit the
enslavement of prisoners, who are still required to work for little or no pay in
various public and private industries. In 2010, a federal court held that
'prisoners have no enforceable right to be paid for their work under the
Constitution.' Yet, across the country, prison labor remains essential to
running prisons and services beyond prison walls. They cook and clean, work in
fields, manufacturing warehouses, and call centers, fight wildfires, do
commercial laundry, make masks and hand sanitizer, sometimes for as little as
two cents an hour — if anything — often under threat of punishment."
https://innocenceproject.org/parchman-farm-prison-mississippi-history/ [889]
In these historical encounters between hardline rulers
and the bete noir substances of their time, we perceive the successful leaders
psychotic, and/or needed to be perceived as psychotic, to instil fear in the
greatest number for reasons which make no sense today, and probably didn't then.
Unlike their papal counterparts, Sultans Mehmet III,
Marud IV and Ibrahim I certainly didn't believe coffee was Satan's drink.
Clement, whose influence on coffee is debated, was
typically motivated by greed and profit. But heresy, not drug use, was the
expedient and well-understood target for the prohibition of the era. With no
antipopes to deal with and a big tourism jubilee to do, coffee got the official
western European theological thumbs up. And the rest is history.
Marijuana convictions, fuelled by the propaganda of a
xenophobic and war-drum-beating media, supply an economical workforce to the
poultry industry.
Is this a credible problem or an appropriate approach
to its solution?
Like Nixon according to Erlichman, it wasn't reallly
about drugs at all.
"In Murad's Istanbul, religious leaders preached on
street corners that coffee would inspire indecent behavior, paralleling the
ZPPPD's blanket appeal to "appropriateness". As the bean moved west into Europe,
physicians rallied against it, claiming that coffee would 'dry up the
cerebrospinal fluid' and cause paralysis."
Thus in England in 1674, The Womens [sic] Petition
Against Coffee spoke of their horror:
"... the Excessive use of that Newfangled, Abominable,
Heathenish Liquor called COFFEE, which Riffling Nature of her Choicest
Treasures, and Drying up the Radical Moisture, has so Eunucht our Husbands that
they are become as unfruitful as those Desarts whence that unhappy Berry is said
to be brought."
This has been debunked as recently as 2018. In 21,403
men aged 40-75:
"No significant differences were identified for
patients with incident ED after comparing highest (≥4 cups/day) with lowest (0
cups/day) categories of total (hazard ratio (HR) = 1.00, 95% confidence interval
(CI): 0.90, 1.11) and regular coffee intakes (HR = 1.00, 95% CI: 0.89, 1.13)."
https://pubmed.ncbi.nlm.nih.gov/29020139/ [887]
But the real concern in the reign of Charles II was
not physiological, but political. And so it did not sound much like impotence
was the problem when a year later, in 1675, a royal edict stated that
coffeehouses "have produced very evil and dangerous effects," and were also a
"disturbance of the peace and quiet realm."
"King Charles II issued an order to shut down all
coffeehouses after he traced some clever but seditious poetry to them. The
backlash was throne-shaking. In just 11 days, Charles reversed his ruling."
"'I think maybe he recalled that they had beheaded his
father,' [author of Uncommon Grounds: The History of Coffee and How It
Transformed Our World, Mark] Pendergrast says. "He didn't want to stir up too
much trouble."
England's newly-restored monarch faced a balancing
act:
"On the one hand, by 1685, the duties collected on the
goods sold by coffee houses amounted to four percent of all of England's neat
excise produce, proving incredibly profitable to the crown. One the other hand,
however, Charles' throne still remained on slippery grounds, and the threat that
the coffeehouses posed was impossible to ignore. Charles was very much aware
that dissenting groups such as Thomas Harrington's republican Rota Club was
meeting a Mile' Coffeehouse in London. There was also a belief among the elite
of London that dissenters were just plain associated with coffeehouse culture in
general, which made them want to squash the threat to the Restoration.
Coffeehouses were thus sometimes suppressed, and duties on the goods they sold
were increased."
However:
"Charles II failure to disrput the coffeehouse culture
of free converse and ideas was an indication of England's ability to progress
out of the shadow of its despotic monarchs and claim civil liberties for the
common person."

Here's a reproduction of a letterbox used at
"Button's Coffee House: Located on Russell Street in
Covent Garden. Also known as The Lion's Head Letter Box because patrons could
drop of pieces of their writing there to be published in the local newspaper The
Guardian."
https://sites.udel.edu/britlitwiki/the-coffeehouse-culture/ [885]
Well we all know how the story ended. Mormons don't
drink coffee. Everyone else can buy coffee, at any age, in any quantity, and the
Guardian still causes trouble occasionally.
Do you think if cannabis had arrived in Slovenia
before other now-legal drugs, the situation might now be reversed?
Which foods and medicines arrive where first is a
matter of a mixture of climatic, evolutionary, historical and geographical
circumstances, would you agree?
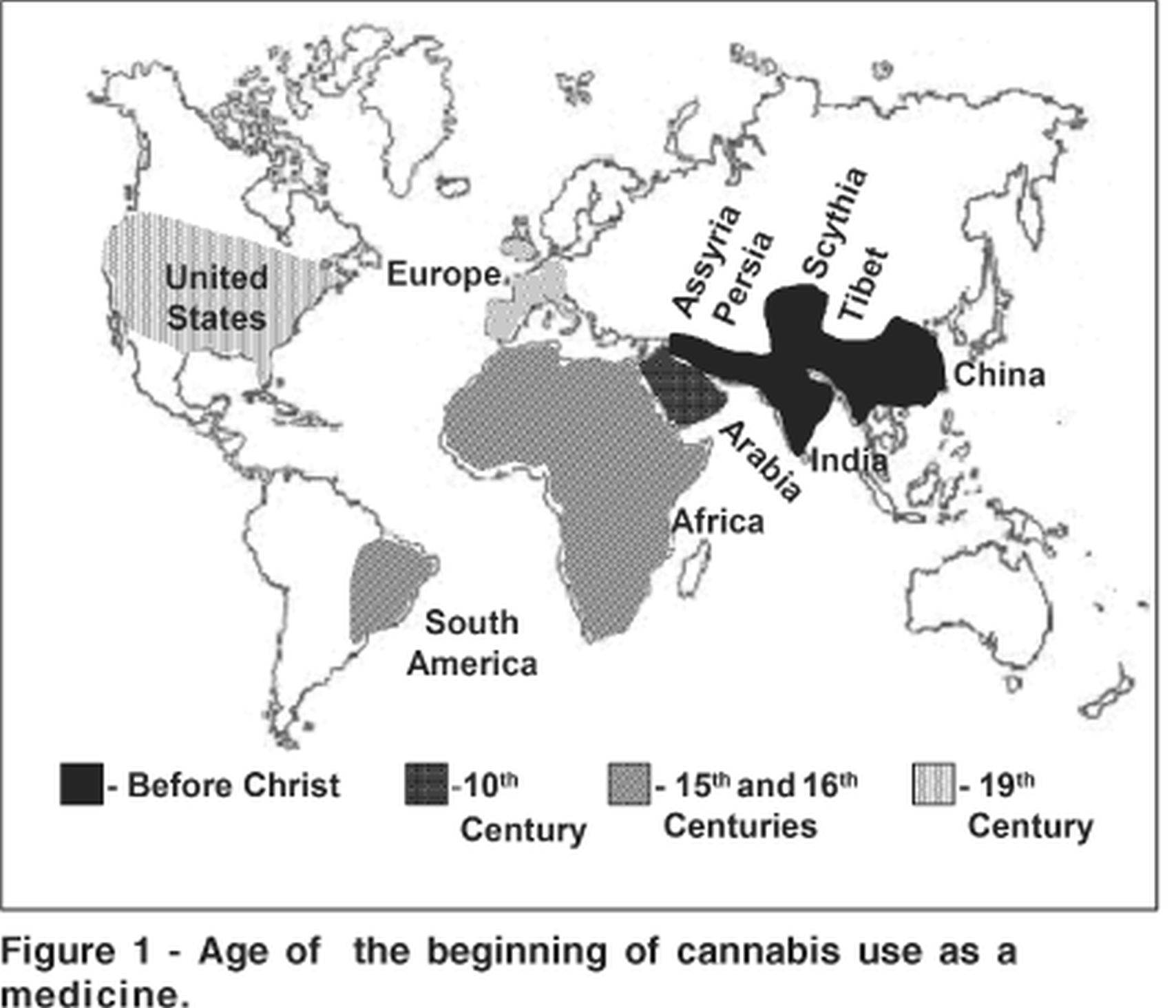
Let's just cannabis had arrived before coffee or
alcohol. Would the authorities today be saying, well, just because we made this
mistake legalising cannabis, it was a very long time ago, and we all use
cannabis now and banning it would be too unpopular?
In this scenario where cannabis came first, wouldn't
the authorities conclude that allowing the newcomers coffee or alcohol would
only be adding to our morbidity, and these should remain illegal and prosecution
used as a source of income for the judiciary and government?
If you knew back then that 40 cups of coffee would
stand a 50% chance of killing you, that would finish legalisation of coffee
completely?
Gang wars might ensue. In 1777, Frederick the Great of
Prussia issued a manifesto claiming beer's superiority over coffee. He argued
that coffee interfered with the country's beer consumption, and royally urged
Prussians to begin the day properly. "His Majesty was brought up on beer," he
proclamation read. Sniffer troops were deployed across Prussia.
"The Kingdom of Yugoslavia ratified the International
Opium Convention on 4 September 1929. The first law to sanction drug abuse was
the Criminal Code of the Kingdom of Serbs, Croats and Slovenes passed on 27
January 1929 and which entered into force on 1 January 1930."
However Wikipedia also advises that King Alexander I
renamed the Kingdom of Serbs, Croats and Slovenes to the Kingdom of Yugoslavia
on 6 January 1929.
So cannabis was first swept into this phoney
"narcotics" grouping 11411 days before the UN Single Convention, and you could
get '"a prison sentence of up to 6 months for "serving" narcotic drugs in the
section "Crimes against Public Health"'
https://en.wikipedia.org/wiki/Cannabis_in_Kosovo [216]
What the law was in the intervening years between 1
January 1930 and 1 January 2000 is somewhat of a mystery to this Defendant.
However it is surely undisputed that continuity existed in some form up to and
after the SCND.
Some of the effects of psychedelics are a kind of
opposite to ADHD, as they enhance traits such as mindfulness and empathy.
Therefore psychedelics like LSD tend to reduce crime because they increase
awareness of the victim, where there is one. In support of these assertions are
examinations of LSD effects in ADHD individuals.
Haijen et al at the Department of Neuropsychology and
Psychopharmacology, Faculty of Psychology and Neuroscience, Maastricht
University, studied "Trait mindfulness and personality characteristics in a
microdosing ADHD sample: a naturalistic prospective survey study" (2023):
"Background: Microdosing (MD), repeatedly taking
psychedelics in small, non-hallucinogenic amounts, has been practiced by
individuals to relieve attention deficit hyperactivity disorder (ADHD) symptoms.
Generally, adults diagnosed with ADHD have lower levels of mindfulness and
differ in personality structure from non-ADHD adults. How MD affects mindfulness
and personality in adults with ADHD remains unexplored.
"Aim: This study aimed to investigate the effects of
4 weeks of MD on mindfulness and personality traits in adults diagnosed with
ADHD and those experiencing severe ADHD symptoms. It was expected that
mindfulness and the personality traits conscientiousness, extraversion,
agreeableness, and openness would increase and neuroticism would decrease after
4 weeks of MD compared to baseline. It was explored if using conventional ADHD
medication alongside MD and/or having comorbidities influenced MD-induced
effects.
"Methods: An online prospective naturalistic design
was used to measure participants before MD initiation and 2 and 4 weeks later.
Validated self-report measures were used assessing mindfulness (15-item Five
Facet Mindfulness Questionnaire) and personality traits (10-item version of the
Big Five Inventory) at three time points.
"Results: The sample included n = 233, n = 66, and
n = 44 participants at the three time points, respectively. Trait mindfulness,
specifically description and non-judging of inner experience, was increased, and
neuroticism was decreased after 4 weeks of MD compared to baseline. The
remaining personality traits remained unchanged. Using conventional medication
and/or having comorbid diagnoses did not change the MD-induced effects on
mindfulness and personality traits after 4 weeks.
"Conclusion: MD induced changes in otherwise stable
traits. Future placebo-controlled studies are warranted to confirm whether these
changes occur in a controlled setting."
They explain:
"Mindfulness can broadly be divided into two
dimensions: self-regulation of attention and acceptance. First, self-regulation
of attention is characterized by attending, observing, and becoming aware of
one’s thoughts, feelings, and sensations without getting caught in ruminative
thought streams. Mindfulness facets that belong to this domain include
observation, description, and acting with awareness. Second, the acceptance
domain involves taking an open, and accepting stance toward one’s observed
experience, thereby inhibiting emotional impulsive responses to whatever is
observed. Mindfulness facets that belong to this domain include non-judging of
inner experience and non-reactivity to inner experience. Generally, individuals
diagnosed with ADHD scored on average lower on trait mindfulness compared to
individuals without an ADHD diagnosis. Interestingly, enhanced levels of
mindfulness have been reported after MD and mindfulness scores were higher in
current and former microdosers compared to MD-naïve controls. Though one
prospective (survey) study investigating adults (excluding individuals with
mood, anxiety, substance use, psychotic or dissociative disorders) did not find
a change in mindfulness scores after MD, and another prospective study
investigating a general population sample could not attribute changes to MD
solely. However, the effects of MD on mindfulness in individuals diagnosed with
ADHD, or individuals experiencing severe ADHD symptoms, have not been
investigated yet."
A bit more about the parameters:
"Mindfulness was strongly and positively related to
conscientiousness (i.e., being well-organized, responsible, and efficient) and
negatively related to neuroticism (i.e., negative affectivity and emotionally
unstable). While the relationships between mindfulness and conscientiousness and
neuroticism have been a consistent finding, the relationships between
mindfulness and the other personality traits agreeableness (i.e., compromising
with, and trusting others), extraversion (i.e., positive emotionality and
socially engaged), and openness (i.e., curious and willing to explore new
experiences) were overall positive, yet less strong. Individuals diagnosed with
ADHD score generally lower on conscientiousness and higher on neuroticism
compared to controls. Though the associations between ADHD and extraversion and
agreeableness are less strong, both traits tend to be lower in ADHD. Openness is
generally unrelated to ADHD. Interestingly, openmindesness (i.e., openness) was
higher and negative emotionality (i.e., neuroticism) was lower in current and
former microdosers compared to MD-naïve controls. Also, a qualitative interview
study reported that participants experienced increases in openness and
extraversion following MD, although no explicit mention of other personality
traits was made."
https://www.frontiersin.org/articles/10.3389/fpsyt.2023.1233585/full [4249]
However inconclusive researchers would like their
results to be, they do not support a hypothesis that microdosing, or normal
doses, of classical psychedelics, reduce conscientiousness towards, or empathy
with, others.
Since these prosocial traits are evidently lacking in
criminal behaviour, the only criminality associated with psychedelics is the
criminality of their criminalization. In short, the ZPPPD is a pro-crime law!
Anyway, the authorities cannot be as bothered about
crime as they claim. Although Spencer (1963) declared in the British Journal of
Psychiatry without further explanation his rather confused opinion that sensory
hyperacuity was of "no therapeutic value" he wrote that LSD was able
"...to bring into consciousness repressed traumatic
experiences and relationships suffered by the patient during childhood, such as
those of parental rejection and hostility, sexual assaults, the sense of
loneliness and impending doom engendered by inhalant anaesthetics."
The mysterious paradox of catharsis unconnected with
consciousness deepened as
"LSD appeared to be unique in the ease with which
under its influence these deeply repressed memories could be recovered, and
analysts using the drug in relatively small doses found that it would produce in
one or two sessions unconscious material which would normally only be recovered
after some months of analysis."
https://scispace.com/papers/permissive-group-therapy-with-lysergic-acid-diethylamide-39q8clxg7v
[5642]
They meant well, but the explanation may simply be
that the "therapists" weren't tripping themselves, with the failure to
communicate being the reason why they were still puzzling over why higher doses
"were also the only doses where a statistically significant reduction in
symptoms after four weeks was found" [5559,
5560]
73 years later.
This particular way of reducing crime soon became a
crime. Possibly psychologists were worried they wouldn't have enough to do, as
the effects on Slovenia's national religion were devastating. Hendricks et al
(2014) were able to find processes by which "Hallucinogen use predicts reduced
recidivism among substance-involved offenders under community corrections
supervision":
"In this longitudinal study, we examined the
relationship between naturalistic hallucinogen use and recidivism among
individuals under community corrections supervision with a history of substance
involvement (n=25,622). We found that hallucinogen use predicted a reduced
likelihood of supervision failure (e.g. noncompliance with legal requirements
including alcohol and other drug use) while controlling for an array of
potential confounding factors (odds ratio (OR)=0.60 (0.46, 0.79)). Our results
suggest that hallucinogens may promote alcohol and other drug abstinence and
prosocial behavior in a population with high rates of recidivism."
https://www.ovid.com/journals/jpsyc/abstract/10.1177/0269881113513851~hallucinogen-use-predicts-reduced-recidivism-among?redirectionsource=fulltextview
[5643]
Worse yet, the patients were threatening to gang
together and dispense with the pointless psychotherapists altogether. According
to Trope et al (2019)
"...participants from recent psychedelic-assisted
individual psychotherapy trials have repeatedly requested to meet other
participants (Bossis 2015; Bradberry et al. 2017) and have attested to the
importance of these connections for corroborating often intense,
difficult-to-describe, psychedelic experiences and reinforcing their beneficial
effects (Bradberry et al. 2017). This echoes the clinical impression of early
researchers that group involvement, whether in mutual aid groups like Alcoholics
Anonymous or formal group therapy, could help solidify therapeutic gains by
extending the intrapsychic experiences of high-dose psychedelic sessions into
the interpersonal relationships of the group setting (Osmond et al. 1967)."
Numerous pre-prohibition studies are cited, for
example one by Sven E Jensen in a Saskatchewan mental hospital based around
Alcoholics Anonymous in 1962, as if alcoholism and mental illness were somehow
connected:
"35 patients received the group therapy and AA
components without the LSD session, and 45 'controls' received only
treatment-as-usual in the hospital during the same time period. Follow-up data
obtained 6-18 months after discharge revealed that in the LSD condition (n=58),
34 remained sober and 7 were considered improved ('drinking definitely less than
before'). Of those who received group components without LSD (n=35), 4 were
sober and 4 were improved. Of the controls (n=45), 7 were sober and 3 improved.
The increased proportion of sobriety or improvement in the LSD condition was
reported as statistically significant compared to the two control arms."
https://escholarship.org/content/qt90n3z820/qt90n3z820.pdf [5644]
Another pre-prohibition study, by Mogar and Savage
(1964), reported "Personality change associated with psychedelic (LSD) therapy":
"MMPI data are presented which were obtained from 60
patients prior to, 2 mo. after, and 6 mo. after a brief psychotherapeutic
program culminating in a single, large dose LSD session. While all sub-groups
show significant positive changes at 2 mo., some patients manifest a tendency
towards regression while others either consolidate initial gains or display
further improvement at 6 mo. The results indicated that the nature, magnitude,
and stability of changes following a psychedelic experience were related to
personality variables, severity of illness and modal defense patterns."
https://psycnet.apa.org/buy/1966-01700-001 [5649]
Of course alcohol cannot cause crime because it is
legal.
Conscientiousness was another threat to crime
proliferation and employment in the justice and analysis industries. Long-term
conscientiousness was elevated with 200 μg of LSD in a 2024 study by Holze et
al:
"This finding aligns with the high proportion of
people in both trials who rated the psychedelic experience as among the most
meaningful experiences in life and is consistent with studies in healthy
volunteers, which also consistently demonstrated enduring positive effects.
Healthy participants attributed positive life changes to their psychedelic
experiences, mirroring findings from therapeutic studies. Notably, research in
healthy participants reports shifts in personality traits, particularly an
increase in the trait openness, but changes in agreeableness and
conscientiousness have also been observed." [3376]
Scientists have even
discovered that tripping in prison might not be the best venue for a
transformational experience. Reporting on psychedelic assisted therapy (PAT) -
elsewhere found to be no more effective than just plain old psychedelics without
the kind of observation through which the dull live vicariously through the more
exciting lives of the less innocent - Tereza Dlestikova (2025) advises that:
"The paper contends that community-based, post-release settings are more
appropriate and ethically defensible for PAT delivery than correctional
institutions. "
https://www.sciencedirect.com/science/article/abs/pii/S0955395925003639
[5646]
Since ADHD often manifests itself in behaviour
resembling an angry toddler, it is pertinent to ask if age and experience, as a
function of heightened depth of awareness, really does bring wisdom. Those of us
who have survived to make such an assessment will be unsurprised to see "Age
increases brain complexity" was confirmed, by Anokhin et al (1996).
"This study investigated age-related changes in the
human brain function using both traditional EEG analysis (power spectra) and the
correlation dimension, a measure reflecting the complexity of EEG dynamics and,
probably, the complexity of neurophysiological processes generating the EEG.
Assuming that the accumulation of individual experience is determined by the
formation of functionally related groups of neurons showing a repetitive
synchronous activation (cell assemblies), an increase in the number of such
independently oscillating cortical cell assemblies can be expected, despite a
decline of some metabolic and memory functions with normal ageing. Thus, the
‘wisdom of old age’ may find its neurophysiological basis in greater complexity
of brain dynamics compared to young ages. The experimental hypothesis was that
EEG dimension steadily increases with age. In order to test this hypothesis the
resting EEGs of 5 age groups from 7 to 60 were analysed. The results confirm the
hypothesis: after a jump in the brain dynamics complexity during puberty a
linear increase with age is observed. During maturation (7–25 years), the
maximum gain in complexity occurs over the frontal associative cortex."
https://www.sciencedirect.com/science/article/abs/pii/0921884X96955733
[4250]
Martin and Nichols (2016) report "Psychedelics Recruit
Multiple Cellular Types and Produce Complex Transcriptional Responses Within the
Brain" and
"...provide evidence that a small subset of
5-HT2A-expressing excitatory neurons is directly activated by psychedelics and
subsequently recruits other select cell types including subpopulations of
inhibitory somatostatin and parvalbumin GABAergic interneurons, as well as
astrocytes, to produce distinct and regional responses. To gather data regarding
the response of specific neuronal populations, we developed methodology for
fluorescence-activated cell sorting (FACS) to segregate and enrich specific
cellular subtypes in the brain. These methods allow for robust neuronal sorting
based on cytoplasmic epitopes followed by downstream nucleic acid analysis,
expanding the utility of FACS in neuroscience research."
https://www.thelancet.com/article/S2352-3964(16)30406-6/fulltext [3723]
"Can Synaptic Loss Explain Cortical Thinning in Schizophrenia?" ask Sapienza et
al (2025):
"Kassem and colleagues, by combining structural MRI and confocal microscopy,
demonstrated that stressed mice showed gray matter losses of 10 and 15% in the
anterior cingulate cortex (ACC) and hippocampus, coupled with a loss of synaptic
spine density of up to 60% but no changes in the number or volumes of the somas
of neurons, astrocytes, or oligodendrocytes. Moreover, there was a strong linear
relationship between dendritic volume loss and MRI-estimated gray matter volume
loss. A similar technique was used by Keifer and colleagues in a study on the
auditory fear-conditioning paradigm in mice to investigate its effects on
different cortical areas. They found increased gray matter voxel intensity in
several brain regions compared to the controls. Focusing on the auditory cortex,
they described concurrent increases in dendritic spine density with a positive
correlation between dendritic spine density and gray matter voxel intensity.
Taken together, these preclinical findings indicate that synaptic changes could
contribute at least partially to cortical thinning and neurostructural
alterations seen in schizophrenia, despite not proving such a relationship."
https://pmc.ncbi.nlm.nih.gov/articles/PMC12469418/ [5561]
Even "Cannabidiol reverses microglia activation and loss of parvalbumin
interneurons and perineuronal nets in a mouse model of schizophrenia". Da Silva
et al (2025) explain:
"Our group has reported that repeated CBD treatment prevented and reversed the
social and cognitive impairments in a mouse model of SCZ based on chronic
treatment with the [glutamate N-methyl-d-aspartate receptor] NMDAR antagonist
MK-801. Also, CBD, similar to the second-generation antipsychotic clozapine,
prevented the decrease in the number of [parvalbumin-positive] PV+ interneurons
in the PFC."
"Most of the PV+ interneurons in the brain are surrounded by the perineuronal
nets (PNNs), a specialized extracellular matrix that aggregates around the
neuronal soma and is directly involved in the regulation of PV+ interneurons
synaptic plasticity, as well as in their protection against oxidative and
metabolic damage. PNNs mature gradually, in an experience-dependent manner,
during the maturation of neural circuits in early adulthood, coinciding with the
age of SCZ onset. Decreases in PNNs, including those surrounding PV+
interneurons, have also been observed in SCZ patients and animal models of the
disease. However, the mechanisms underlying PNN deficits in SCZ, which may
include the functional capacity of microglial cells to remodel the brain
extracellular matrix, are still unclear."
It does not sound as if the Universities of Sao Paulo and Saarland are doing the
sort of thing for which, in Hong Kong, they might be fined HK$1 million and get
seven years in jail. But challenges remain:
"The protective effect of CBD on PV+ interneurons seem to depend on 5-HT1A and
CB2 receptors in the vHIP [ventral hippocampus] but not in the prelimbic mPFC.
At the moment, we have no explanation for this difference. A study with local
CBD microinjection into the prelimbic cortex indicated that 5-HT1A receptors
play a complex role in this structure. CBD could produce anxiolytic or
anxiogenic responses, depending on previous exposure to restraint stress and
glucocorticoid levels. Both responses, however, were blocked by [5-HT1AR
receptor antagonist and potent dopamine 4 receptor (D4R) full agonist]
WAY100635. Further studies are needed to understand the role of these receptors
and the mechanisms of the PV+/PNNs CBD protective effect in this region."
https://www.biorxiv.org/content/10.1101/2024.10.21.619352v1.full.pdf [5339]
Returning to where we began in this section, the 2023
ADHD paper of Ittiphakorn et al...
"Patients were identified from the UK Medical Cannabis
Registry. Primary outcomes were changes in the following patient-reported
outcome measures (PROMs) at 1, 3, 6, and 12 months from baseline: EQ-5D-5L index
value, generalized anxiety disorder-7 (GAD-7) questionnaire, and the single-item
sleep quality score (SQS). Secondary outcomes assessed the incidence of adverse
events. Statistical significance was defined as p < 0.050.
"Results
Sixty-eight patients met the inclusion criteria.
Significant improvements were identified in general HRQoL [health-related
quality of life] assessed by EQ-5D-5L index value at 1, 3, and 6 months
(p < 0.050). Improvements were also identified in GAD-7 [generalized anxiety
disorder-7 questionnaire] and SQS [sleep quality score] scores at 1, 3, 6, and
12 months (p < 0.010). 61 (89.71%) adverse events were recorded by 11 (16.18%)
participants, of which most were moderate (n = 26, 38.24%)." [4264]
Another HRQoL-based study looked at medical cannabis
patients with primary and secondary diagnoses including agoraphobia, anxiety,
attention-deficit/hyperactivity disorder, autistic spectrum disorder, cancer
pain, chemotherapy-induced nausea and vomiting, chronic non-cancer pain, cluster
headaches, complex regional pain syndrome, Crohn's disease, depression, eating
disorder, Ehlers-Danlos Syndrome, endometriosis, epilepsy adult, fibromyalgia,
headache, inflammatory arthritis, insomnia, migraine, multiple sclerosis,
neuropathic pain, obsessive-compulsive disorder, osteoarthritis, palliative
care, Parkinson's, post-traumatic stress disorder, rare and challenging skin
condition, Tourette's syndrome, trigeminal neuralgia, and ulcerative colitis.
"Results: 1378 patients prescribed Adven® CBMPs
(Curaleaf International, Guernsey, UK) were included in the final analysis. 581
(42.16%) participants were current users of cannabis at baseline. 641 (46.51%),
235 (17.05%), and 502 (36.43%) patients were treated with oils, dried flowers,
or a combination of the two, respectively. Improvements were found in all PROMs
in each route of administration at 1, 3, 6, and 12 months from baseline (p <
0.010). Those prescribed dried flower only or both oils and dried flower
experienced greater improvements in GAD-7, SQS, and EQ-5D-5L index values at 12
months (p < 0.050). There was no difference in outcomes between those prescribed
dried flower only or dried flower with oils (p > 0.050). 3663 (265.82%) adverse
events were reported by 297 (21.55%) patients.
"Conclusion: There was an associated improvement in
self-reported anxiety, sleep quality, and HRQoL in patients treated with the
CBMPs. Those prescribed treatment formulations including dried flower were most
likely to show a clinical improvement. However, these results must be
interpreted with caution given the limitations of study design."
https://onlinelibrary.wiley.com/doi/pdfdirect/10.1002/npr2.12403 [4292]
Murphy et al in "A cohort study comparing the effects
of medical cannabis for anxiety patients with and without comorbid sleep
disturbance" (2023) continues with science's astounding discoveries:
"Background: Research on cannabis- based medicinal
products (CBMPs) in anxiety remains inconclusive due to a paucity of
high-quality evidence. Studies indicate a bidirectional relationship between
generalized anxiety disorder (GAD) and sleep disruption, but it is unclear how
this affects CBMP treatment outcomes. This study aims to compare the patient-
reported outcome measures (PROMs) of patients prescribed CBMPs for GAD, with and
without impaired sleep.
"Methods: Changes in PROMs were recorded from baseline
to 1, 3, 6, and 12 months between those with impaired or unimpaired sleep.
Multivariate logistic regression was applied to compare factors associated with
a clinically significant improvement in GAD-7 at 12 months. Secondary outcomes
included adverse event incidence and frequency.
"Results: Of the 302 patients that fit the inclusion
criteria, mean GAD-7, single-item sleep quality, and EQ-5D-5L index values
improved at all time points (p< 0.001). A relationship between sleep impairment
and clinically significant changes in GAD-7 at 1 and 3 months was identified (p
≤ 0.01). On multivariate regression, only baseline GAD severity was associated
with an increased likelihood of observing a clinically significant improvement
in anxiety (p< 0.001). Seven hundred and seven (234%) adverse events were
reported by 55 (18.21%) participants.
"Conclusions: This study observed an association
between CBMP treatment and improvements in anxiety in patients with GAD. While
patients with comorbid sleep dis-ruption had greater improvements in anxiety,
the differences were not maintained in a multivariate analysis. Baseline anxiety
severity may be a predictor for CBMP treatment outcomes"
https://onlinelibrary.wiley.com/doi/epdf/10.1002/npr2.12407 [4301]
As for the role of NECUD in mental health, a 2024
study "Youth cannabis use and subsequent health service use for mood and anxiety
disorders: A population-based cohort study" from Toronto
"This study found a modest association between less
than weekly cannabis use and subsequent MAD health service use during youth, but
no evidence of a linear dose-response relationship as weekly or more cannabis
use was not associated with MAD health service use. Sensitivity analyses
suggested that these findings were robust to several different model conditions
while interaction analyses did not find any sex or age differences either on the
multiplicative or additive scale."
But of course the authors could easily have drawn a
line between 1.48 (<weekly) and 0.92 (weekly+), illustrating a linear
relationship between more committed users, and NECUD-afflicted users of not
enough cannabis - consisting of the doubters, the people who only smoke when
they're drunk, the people who only smoke other people's, and whatever is today's
equivalent to what we once described as "weekend hippies". The problem is not
with the existence of a linear relationship, but that the line is going in the
opposite direction to the one McDonald et al expected. Hence their claim there
is no linear relationship at all.
https://www.sciencedirect.com/science/article/abs/pii/S0165178123006443?dgcid=author
[4313]
What can we discover about the real world results of
teenage drug use on educational attainment? The findings of Levola et al from
Helsinki, in which "Adolescent alcohol and cannabis use and early adulthood
educational attainment in the 1986 Northern Finland birth cohort study" were
assessed in 15-16 year olds were rechecked at age 33, a 17 year follow-up.
"Using weighted multivariable models, we examined
prospective associations between age at first drink (AFD), age at first
intoxication (AFI), frequency of alcohol intoxication, as well as self-reported
alcohol tolerance (i.e., number of drinks needed for the subjective experience
of intoxication), and lifetime cannabis use at age 15/16 years with subsequent
educational attainment obtained from comprehensive registers until age 33 in the
Northern Finland Birth Cohort 1986 (6,564 individuals, 49.1% male). Confounding
variables including sex, family structure (intact vs. non-intact), maternal and
paternal education level, behavioural/emotional problems in school at age 7/8
years, having a history of illicit substance use in adolescence, having any
psychiatric diagnosis before age 16, and parental psychiatric diagnoses, were
adjusted for."
and
"Frequent alcohol intoxication (more than three times
in past 30 days) at age 15/16 years (OR 2.00; 95% CI 1.15– 3.48; p=0.014) and
high self-reported alcohol tolerance (<9 alcohol units for males, <7 for
females; OR 2.10; 95% CI 1.21–3.65; p=0.009) were associated with increased ORs
of completing primary education only compared to secondary education (Table 2;
model 1, blocks 3c and 3d). AFI (OR 0.70; 95% CI 0.60–0.82; p<0.001), alcohol
intoxication frequency during the past month at age 15/16 (1–2 times OR 0.78;
95% CI 0.67–0.92; p=0.002; 3 or more times OR 0.42; 95% CI 0.32–0.56; p<0.001)
and high self-reported alcohol tolerance (OR 0.40; 95% CI 0.30–0.52 p<0.001)
were associated with decreased ORs for college/university education compared to
secondary education (Table 2; model 1, blocks 3b, 3c and 3d). Notably, also
those reporting lower alcohol tolerance were less likely to complete
college/university education compared to secondary education when compared to
those with no alcohol use or never been intoxicated (OR 0.85; 95% CI 0.73–0.99;
p=0.038).
"Cannabis use by age 15/16 years was not associated
with of completing primary education only compared to secondary education.
Lifetime use of cannabis or other illicit substance use (yes/no) was associated
with lower odds of college/university education compared to secondary education
(OR 0.68; 95% CI 0.56–0.82; p<0.001) when sex, behavioural/emtional problems at
age 7/8 years and psychiatric diagnoses by age 15/16 years were controlled for
(Table 2: model 1, block 1). However, when family-related variables and
frequency of alcohol intoxication at age 15/16 years were controlled for,
cannabis use was no longer statistically significantly associated with lower
odds of college/university education compared to secondary education (1–4 times
OR 1.03; 95% CI 0.71–1.50; p=0.871; 5 times or more OR 0.88; 95% CI 0.36–2.17;
p=0.780). (Table 2: mode 2, block 3)."
So reading between the lines, while it looked like
smoking in your teens might have something to do with not going on to further
education, once the disruption caused by prohibition - arguing with your parents
and having to leave home because of the prejudices they have been trained by
society to have against their children - once these factors were taken into
account, the association disappeared, coming nowhere near statistical
significance in this large-scale, long term analysis.
"Discussion: In this large birth cohort study with a
17-year followup, younger age at first intoxication, higher frequency of alcohol
intoxication, and high self-reported alcohol tolerance at age 15/16 years were
associated with poorer educational outcomes by the age of 33 years. These
adverse associations were evident regardless of a range of potential
confounders, such as behavioural/ emotional problems at age 7/8 years and
parental education level. The association between adolescent lifetime cannabis
use and educational attainment in adulthood was no longer statistically
significant after adjusting for potential confounders including alcohol use."
https://bmcpublichealth.biomedcentral.com/counter/pdf/10.1186/s12889-024-17693-w.pdf
[4461]
It might not matter either way. Coley et al (2024) in
JAMA Pediatrics
"...drew from data in 47 states, looking at responses
from 898,271 teens. 'With parent consent,' authors explained, 'students from
ninth to twelfth grade self-reported prior month use of cannabis, alcohol,
cigarettes, and e-cigarettes.'
"Passage of recreational cannabis laws (RCL) 'was not
associated with adolescents’ likelihood or frequency of cannabis use,' the
analysis found, 'although negative total effect estimates indicated
significantly lowered use following RCL.' Nor were increases associated with the
launch of recreational cannabis retail sales (RCR)."
and
"this study is the first to evaluate associations
between RCL and RCR policies and adolescent substance use through 2021.”
The results, say the Boston College and the University
of Maryland at College Park team
"...suggest that legalization and greater control over
cannabis markets have not facilitated adolescents’ entry into substance use."
https://jamanetwork.com/journals/jamapediatrics/article-abstract/2817566
[4549]
So in case the Court was considering balancing the
benefits to the over 65s against the rather exaggerated risks of teenage use
these are some factors to take into account.
We show elsewhere that Covid is associated with
gliosis and that cannabis use reduces gliosis. Further evidence in support of
the Article 2 challenge includes "SARS-CoV-2 is associated with changes in brain
structure in UK Biobank" by Douaud et al (2022), who
"...investigated brain changes in 785 participants of
UK Biobank (aged 51–81 years) who were imaged twice using magnetic resonance
imaging, including 401 cases who tested positive for infection with SARS-CoV-2
between their two scans—with 141 days on average separating their diagnosis and
the second scan—as well as 384 controls. The availability of pre-infection
imaging data reduces the likelihood of pre-existing risk factors being
misinterpreted as disease effects. We identified significant longitudinal
effects when comparing the two groups, including (1) a greater reduction in grey
matter thickness and tissue contrast in the orbitofrontal cortex and
parahippocampal gyrus; (2) greater changes in markers of tissue damage in
regions that are functionally connected to the primary olfactory cortex; and (3)
a greater reduction in global brain size in the SARS-CoV-2 cases. The
participants who were infected with SARS-CoV-2 also showed on average a greater
cognitive decline between the two time points. Importantly, these imaging and
cognitive longitudinal effects were still observed after excluding the 15
patients who had been hospitalised. These mainly limbic brain imaging results
may be the in vivo hallmarks of a degenerative spread of the disease through
olfactory pathways, of neuroinflammatory events, or of the loss of sensory input
due to anosmia."
https://www.nature.com/articles/s41586-022-04569-5 [4544]
The infections which can lead to brain loss extend
beyond Covid, according to "Proteomics identifies potential immunological
drivers of postinfection brain atrophy and cognitive decline" by Duggan et al
(2024), who looked at "influenza, pneumonia, tuberculosis, candidiasis/fungal,
miscellaneous bacterial infections, gastrointestinal infections, sexually
transmitted infections, human herpes virus (HHV) infections, viral hepatitis,
miscellaneous viral infections, upper respiratory tract infections (URTIs),
lower respiratory tract infections (LRTIs), skin and subcutaneous infections,
urinary tract infections, and ‘other’ infections (Supplementary Table 1)."
"Systemic infections may influence dementia risk and
neurodegeneration by triggering an acute inflammatory response or reshaping the
host immune system, as in the case of chronic inflammation. In response to
immune insults, such as pathogens and tissue damage, changes in circulating
inflammatory proteins can influence brain health through a variety of
mechanisms, including their interactions with target cells in the central
nervous system (CNS). For example, elevated cytokine signaling after SARS-CoV-2
infection can result in neuroinflammation and post-acute sequelae despite its
low or absent copy numbers in the CNS. Increases in plasma inflammatory markers
among cognitively normal adults are associated with reduced brain volumes and
cognitive performance, and greater dementia risk decades later. Although several
studies have tied select immune markers (for example, tumor necrosis factor
(TNF), interleukin (IL)-1β) to preceding inflammatory events and ensuing
cognitive performance, it remains unknown how infections relate to an array of
immunological proteins, and which of these proteins may predict changes in brain
regions vulnerable to infection-specific atrophy."
In their sample:
"Less than half (42.9%; n = 421) of participants
exhibited a history of no infection diagnoses and 10.1% (n = 108) exhibited a
history of two or more infection diagnoses. Primary regions of interest (ROIs)
included total brain, gray matter, white matter and lobar volumes, as well as an
AD-signature region volume. Follow-up analyses were performed on lobar
white/gray matter volumes if an infection was significantly associated with a
primary ROI."
And...
"Of the 15 infections examined, 6 were associated with
accelerated brain volume loss, predominantly in temporal gray and/or white
matter regions (Fig. 2 and Supplementary Table 4). Follow-up analyses revealed
that influenza-related volume loss was specific to temporal and occipital lobe
gray matter (Fig. 2a and Extended Data Fig. 1a). Consistent with published
findings, declines in white matter volumes linked to herpetic infections (Fig.
2b) were localized to the temporal lobe (Extended Data Fig. 1b). Accelerated
atrophy in the temporal lobe tied to miscellaneous viral infections was specific
to gray matter (Fig. 2c and Extended Data Fig. 1c). URTIs were similarly linked
to accelerated loss in total temporal lobe volume, but such decreases were not
specific to either white or gray matter (Fig. 2d and Extended Data Fig. 1d).
LRTI-related volume loss in the temporal lobe was exclusive to white matter,
whereas decreases in the occipital lobe were evident in both white and gray
matter (Fig. 2e and Extended Data Fig. 1e). Along with reduced total brain
volume, the gray matter atrophy related to skin and subcutaneous infections was
localized to the temporal and occipital lobes (Fig. 2f and Extended Data Fig.
1f). Although associations between infection history and brain volume changes
were no longer statically significant after false recovery rate (FDR)
correction, such correction may not be appropriate because of the inherent
interdependence of the outcomes (for example, total gray matter loss is related
to total brain volume loss). A history of any of the examined infections and
total frequency of infections were not associated with volume changes. Despite
limited power, sensitivity analyses restricting the comparison group to
participants with a history of no infections (for example, influenza versus no
history of any infection) showed consistent atrophy linked to influenza, herpes
viruses, miscellaneous viral infections and LRTIs, with URTI- and skin and
subcutaneous infection-related atrophy remaining marginally significant
(P ≤ 0.10) (Supplementary Table 4). For primary ROIs, we did not find
differences at baseline and results were similar after adjusting for total
infection frequency (Supplementary Tables 4 and 5). These findings suggest that
specific infections may be associated with accelerated brain volume loss,
particularly in temporal regions."
https://www.nature.com/articles/s43587-024-00682-4 [3379]
In 2025 Pitakbut and Kayser reported that:
"After cannabinoids exhibited promising inhibitory
activity against bacterial (C. perfringens) neuraminidase in a prior experiment,
the authors further evaluated the anti-neuraminidase activity against the
influenza A virus (IAV)....Here, the authors studied nine cannabinoid
derivatives. Figure 4 shows six active cannabinoids, THC, CBD, CBE, CBG, CBL,
and CBN, completely inhibiting IAV neuraminidase activity (100% inhibition)." [4854]
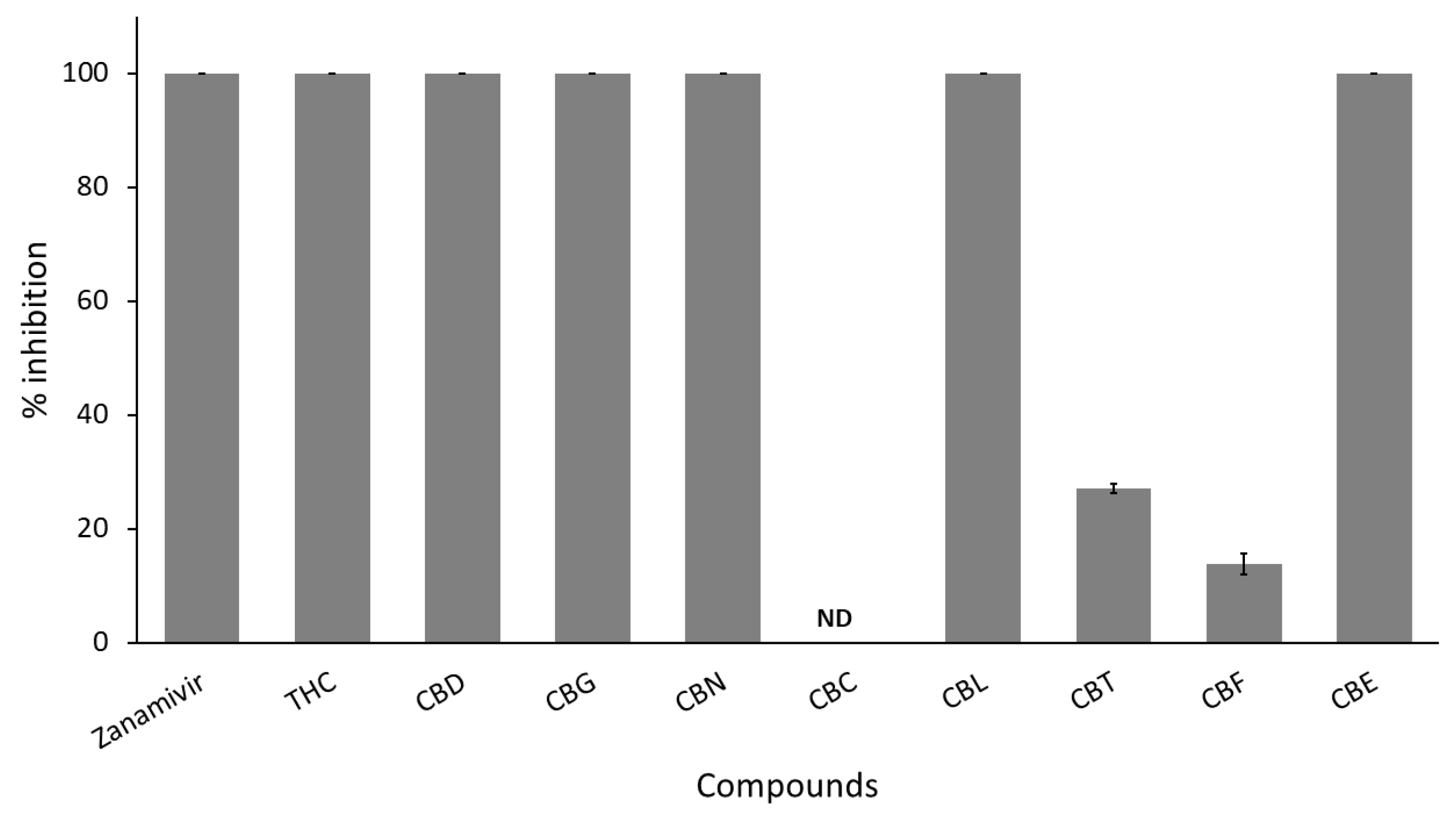
It does not seem likely, therefore, that taking
cannabis away will create an improved medical outcome for the future, present,
or past sufferer from a Covid infection. On the contrary, the evidence shows the
Police action at the time of the pandemic was an anti-health manoeuvre.
"Lifetime Cannabis Use
Is Associated with Brain Volume and Cognitive Function in Middle-Aged and Older
Adults", report Guha et al (2025):
"Using data from the UK Biobank, which includes health information from over
500,000 adults, associations between cannabis use, regional brain volume, and
cognition in participants aged 40-70 years (mean age = 54.5) were evaluated.
Results: Lifetime cannabis use was positively associated with regional brain
volume in CB1-rich regions, including the caudate, putamen, hippocampus, and
amygdala. Greater lifetime use was also linked to better performance in
learning, processing speed, and short-term memory. Individuals reporting use
limited to adolescence also showed larger regional volumes and better cognitive
performance than non-users. Sex differences in cannabis effects on brain volume
and cognition were also observed."
https://pubmed.ncbi.nlm.nih.gov/41379083/ [5718]
----------------------------------------------------------------------------
The Englishman stands
for the rights of everyone disadvantaged, discriminated against, persecuted, and
prosecuted on the false or absent bases of prohibition, and also believes the
victims of these officially-sanctioned prejudices have been appallingly treated
and should be pardoned and compensated.
The Englishman requests the return of his CaPs and other rightful property, for
whose distraint Slovenia has proffered no credible excuse or cause.
The Benedictions represent both empirical entities as well as beliefs. Beliefs
which the Defence evidence shows may be reasonably and earnestly held about the
positive benefits of CaPs at the population level, in which the good
overwhelmingly outweighs the bad. Below, the latest version of this dynamic
list.
THE BENEDICTIONS
REFERENCES
TIMELINE OF DRUG LAW v. SCIENCE